Antibiotic resistance in Pseudomonas aeruginosa remains one of the most challenging phenomena of everyday medical science. The universal spread of high-risk clones of multidrug-resistant/extensively drug-resistant (MDR/XDR) clinical P. aeruginosa has become a public health threat. The P. aeruginosa bacteria exhibits remarkable genome plasticity that utilizes highly acquired and intrinsic resistance mechanisms to counter most antibiotic challenges. In addition, the adaptive antibiotic resistance of P. aeruginosa, including biofilm-mediated resistance and the formation of multidrug-tolerant persisted cells, are accountable for recalcitrance and relapse of infections.
- drug-resistant
- public health concern
- antibiotic stewardship
1. Introduction
2. Inherent Resistome
3. Mutational Resistome
| Responsible Genes | Mechanism of Resistance | Associated Antibiotics |
|---|---|---|
| gyrA | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| gyrB | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| parC | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| parE | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| phoQ, cprS, colR, colS, pmrA, pmrB | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| parR | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexXY) | Aminoglycosides, cefepime | |
| parS | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Hyperproduction of efflux-mediated genes (MexXY) | Fluoroquinolones | |
| mexR | Hyperproduction of efflux-mediated genes (MexAB-OprM) | Fluoroquinolones |
| nfxB | Hyperproduction of efflux-mediated genes (MexCD-OprJ) | Fluoroquinolones, cefepime |
| mexS | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| Downregulation of OprD | Imipenem, meropenem | |
| mexT | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| cmrA, mvaT, PA3271 | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| mexZ, PA5471.1, amgS | Hyperproduction of efflux-mediated genes (MexXY) | MexXY hyperproduction |
| oprD | Inactivation of Porin channels | Imipenem, meropenem |
| ampD, ampDh2, ampDh3, ampR, dacB, mpl | Hyperproduction of AmpC | Ceftazidime, cefepime, piperacillin–tazobactam |
| ftsI | Target modification (PBP3) | Ceftazidime, cefepime, piperacillin–tazobactam |
| fusA1 | Target modification of Aminoglycoside (elongation factor G) | Aminoglycosides |
| glpT | Transporter protein inactivation GlpT | Fosfomycin |
| rpoB | Rifampin target modification, RNA polymerase β-chain | Rifampin |
4. Horizontally Acquired Resistome
5. Antibiotic Resistance by SOS Response
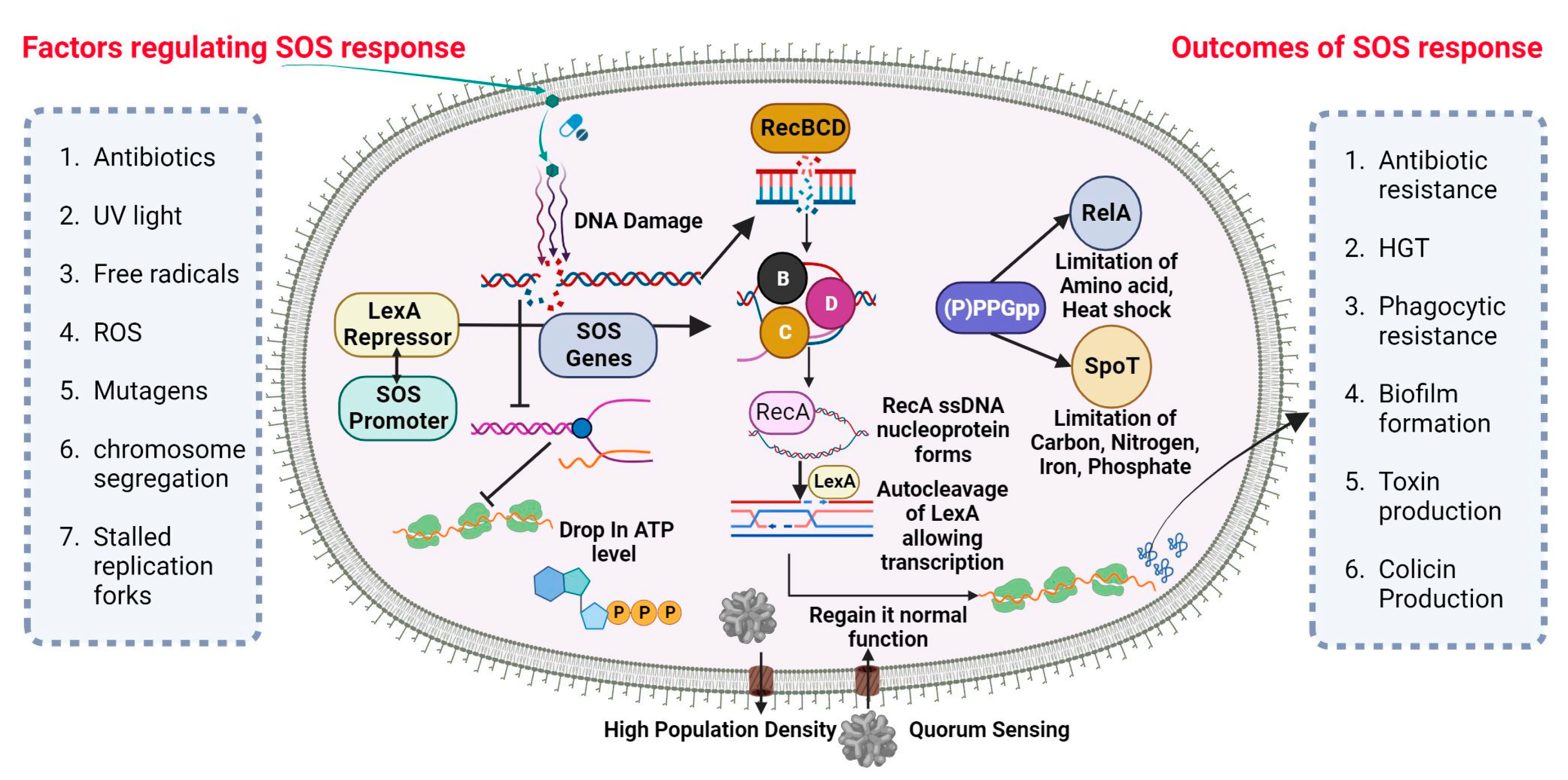
SOS-Dependent Mutagenesis and Resistance
6. Biofilm-Mediated Resistome
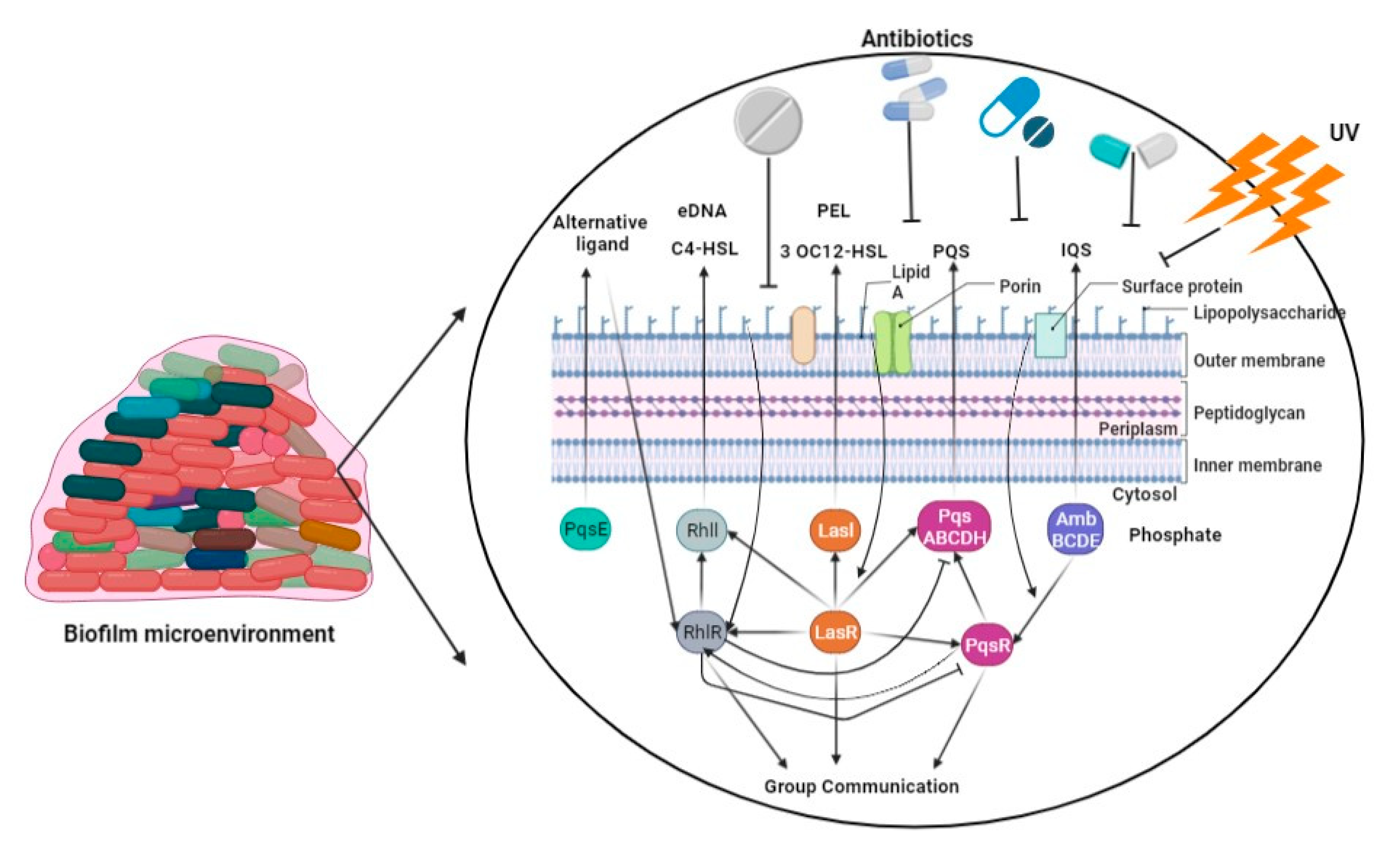
This entry is adapted from the peer-reviewed paper 10.3390/ph16091230
References
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426.
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21–22, 41–59.
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327.
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610.
- Skiada, A.; Markogiannakis, A.; Plachouras, D.; Daikos, G.L. Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2011, 37, 187–193.
- Muller, C.; Plésiat, P.; Jeannot, K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 55, 1211–1221.
- Juan, C.; Torrens, G.; González-Nicolau, M.; Oliver, A. Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol. Rev. 2017, 41, 781–815.
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19.
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical Characterization of the Naturally Occurring Oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048.
- Fajardo, A.; Hernando-Amado, S.; Oliver, A.; Ball, G.; Filloux, A.; Martinez, J.L. Characterization of a novel Zn2+-dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2972–2978.
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418.
- Cabot, G.; Ocampo-Sosa, A.A.; Tubau, F.; Macia, M.D.; Rodríguez, C.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; Martínez-Martínez, L.; et al. Overexpression of AmpC and Efflux Pumps in Pseudomonas aeruginosa Isolates from Bloodstream Infections: Prevalence and Impact on Resistance in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 2011, 55, 1906–1911.
- Moya, B.; Dötsch, A.; Juan, C.; Blázquez, J.; Zamorano, L.; Haussler, S.; Oliver, A. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009, 5, e1000353.
- Cabot, G.; Bruchmann, S.; Mulet, X.; Zamorano, L.; Moyà, B.; Juan, C.; Haussler, S.; Oliver, A. Pseudomonas aeruginosa Ceftolozane-Tazobactam Resistance Development Requires Multiple Mutations Leading to Overexpression and Structural Modification of AmpC. Antimicrob. Agents Chemother. 2014, 58, 3091–3099.
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Martín-Pena, M.L.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663.
- Lahiri, S.D.; Alm, R.A. Identification of Novel VEB β-Lactamase Enzymes and Their Impact on Avibactam Inhibition. Antimicrob. Agents Chemother. 2016, 60, 3183–3186.
- Haidar, G.; Philips, N.J.; Shields, R.K.; Snyder, D.; Cheng, S.; Potoski, B.A.; Doi, Y.; Hao, B.; Press, E.G.; Cooper, V.S.; et al. Ceftolozane-Tazobactam for the Treatment of Multi-drug-Resistant Pseudomonas aeruginosa Infections: Clinical Effectiveness and Evolution of Resistance. Clin. Infect. Dis. 2017, 65, 110–120.
- Berrazeg, M.; Jeannot, K.; Enguéné, V.Y.N.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-Lactamase AmpC Increase Resistance of Pseudomonas aeruginosa Isolates to Antipseudomonal Cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255.
- Caballero, J.D.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.W.; Waters, V.J.; Hwang, D.M.; et al. Selective Sweeps and Parallel Pathoadaptation Drive Pseudomonas aeruginosa Evolution in the Cystic Fibrosis Lung. mBio 2015, 6, e00981-15.
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S.; et al. Evolution of the Pseudomonas aeru-ginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 2017, 7, 5555.
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; et al. Deciphering the Resistome of the Widespread Pseudomonas aeruginosa Sequence Type 175 International High-Risk Clone through Whole-Genome Sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423.
- Del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S.; et al. Genomics and Susceptibility Profiles of Extensively Drug-Resistant Pseudomonas aeruginosa Isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17.
- Cabot, G.; Zamorano, L.; Moyà, B.; Juan, C.; Navas, A.; Blázquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa Antimicrobial Resistance and Fitness under Low and High Mutation Rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778.
- Cabot, G.; Florit-Mendoza, L.; Sánchez-Diener, I.; Zamorano, L.; Oliver, A. Deciphering β-lactamase-independent β-lactam resistance evolution trajectories in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 3322–3331.
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of sidero-phore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007.
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778.
- Hocquet, D.; Berthelot, P.; Roussel-Delvallez, M.; Favre, R.; Jeannot, K.; Bajolet, O.; Marty, N.; Grattard, F.; Mariani-Kurkdjian, P.; Bingen, E.; et al. Pseudomonas aeruginosa may accumulate drug resistance mechanisms without losing its ability to cause bloodstream infections. Antimicrob. Agents Chemother. 2007, 51, 3531–3536.
- Solé, M.; Fabrega, A.; Cobos-Trigueros, N.; Zamorano, L.; Ferrer-Navarro, M.; Ballesté-Delpierre, C.; Reustle, A.; Castro, P.; Nicolás, J.M.; Oliver, A.; et al. In vivo evolution of resistance of Pseudomonas aeruginosa strains isolated from patients admitted to an intensive care unit: Mechanisms of resistance and antimicrobial exposure. J. Antimicrob. Chemother. 2015, 70, 3004–3013.
- Riera, E.; Cabot, G.; Mulet, X.; García-Castillo, M.; del Campo, R.; Juan, C.; Cantón, R.; Oliver, A. Pseudomonas aeruginosa carbapenem resistance mechanisms in Spain: Impact on the activity of imipenem, meropenem and doripenem. J. Antimicrob. Chemother. 2011, 66, 2022–2027.
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228.
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305.
- Mulet, X.; Moyá, B.; Juan, C.; Macià, M.D.; Pérez, J.L.; Blázquez, J.; Oliver, A. Antagonistic Interactions of Pseudomonas aeruginosa Antibiotic Resistance Mechanisms in Planktonic but Not Biofilm Growth. Antimicrob. Agents Chemother. 2011, 55, 4560–4568.
- Bruchmann, S.; Dötsch, A.; Nouri, B.; Chaberny, I.F.; Häussler, S. Quantitative Contributions of Target Alteration and Decreased Drug Accumulation to Pseudomonas aeruginosa Fluoroquinolone Resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368.
- Feng, Y.; Jonker, M.J.; Moustakas, I.; Brul, S.; ter Kuile, B.H. Dynamics of Mutations during Development of Resistance by Pseudomonas aeruginosa against Five Antibiotics. Antimicrob. Agents Chemother. 2016, 60, 4229–4236.
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685.
- Greipel, L.; Fischer, S.; Klockgether, J.; Dorda, M.; Mielke, S.; Wiehlmann, L.; Cramer, N.; Tümmler, B. Molecular Epidemiology of Mutations in Antimicrobial Resistance Loci of Pseudomonas aeruginosa Isolates from Airways of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2016, 60, 6726–6734.
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643.
- Gutu, A.D.; Sgambati, N.; Strasbourger, P.; Brannon, M.K.; Jacobs, M.A.; Haugen, E.; Kaul, R.K.; Johansen, H.K.; Høiby, N.; Moskowitz, S.M. Polymyxin Resistance of Pseudomonas aeruginosa phoQ Mutants Is Dependent on Additional Two-Component Regulatory Systems. Antimicrob. Agents Chemother. 2013, 57, 2204–2215.
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crécy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005, 3, e176.
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651.
- Panda, A.; Drancourt, M.; Tuller, T.; Pontarotti, P. Genome-wide analysis of horizontally acquired genes in the genus Mycobacterium. Sci. Rep. 2018, 8, 14817.
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.P. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92.
- Kothari, A.; Kumar, P.; Gaurav, A.; Kaushal, K.; Pandey, A.; Yadav, S.R.M.; Jain, N.; Omar, B.J. Association of antibiotics and heavy metal arsenic to horizontal gene transfer from multidrug-resistant clinical strains to antibiotic-sensitive environmental strains. J. Hazard Mater. 2023, 443, 130260.
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132.
- Patel, G.; Bonomo, R.A. Status report on carbapenemases: Challenges and prospects. Expert Rev. Anti-Infect. Ther. 2011, 9, 555–570.
- Juan, C.; Conejo, M.C.; Tormo, N.; Gimeno, C.; Pascual, Á.; Oliver, A. Challenges for accurate susceptibility testing, detection and interpretation of β-lactam resistance phenotypes in Pseudomonas aeruginosa: Results from a Spanish multicentre study. J. Anti-Microb. Chemother. 2013, 68, 619–630.
- Botelho, J.; Grosso, F.; Quinteira, S.; Mabrouk, A.; Peixe, L. The complete nucleotide sequence of an IncP-2 megaplasmid unveils a mosaic architecture comprising a putative novel blaVIM-2-harbouring transposon in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2017, 72, 2225–2229.
- Van der Zee, A.; Kraak, W.B.; Burggraaf, A.; Goessens, W.H.F.; Pirovano, W.; Ossewaarde, J.M.; Tommassen, J. Spread of Carbapenem Resistance by Transposition and Conjugation Among Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 2057.
- Botelho, J.; Grosso, F.; Peixe, L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J. Antimicrob. Chemother. 2018, 73, 77–83.
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585.
- Del Barrio-Tofiño, E.; Zamorano, L.; Cortes-Lara, S.; López-Causapé, C.; Sánchez-Diener, I.; Cabot, G.; Bou, G.; Martínez-Martínez, L.; Oliver, A.; Galán, F.; et al. Spanish nationwide survey on Pseudomonas aeruginosa antimicrobial resistance mechanisms and epidemiology. J. Antimicrob. Chemother. 2019, 74, 1825–1835.
- Chávez-Jacobo, V.M.; Hernández-Ramírez, K.C.; Romo-Rodríguez, P.; Pérez-Gallardo, R.V.; Campos-García, J.; Gutiérrez-Corona, J.F.; García-Merinos, J.P.; Meza-Carmen, V.; Silva-Sánchez, J.; Ramírez-Díaz, M.I. CrpP Is a Novel Ciprofloxacin-Modifying Enzyme Encoded by the Pseudomonas aeruginosa pUM505 Plasmid. Antimicrob. Agents Chemother. 2018, 62, e02629-17.
- Moya, B.; Zamorano, L.; Juan, C.; Pérez, J.L.; Ge, Y.; Oliver, A. Activity of a new cephalosporin, CXA-101 (FR264205), against be-ta-lactam-resistant Pseudomonas aeruginosa mutants selected in vitro and after antipseudomonal treatment of intensive care unit patients. Antimicrob. Agents Chemother. 2010, 54, 1213–1217.
- Torrens, G.; Cabot, G.; Ocampo-Sosa, A.A.; Conejo, M.C.; Zamorano, L.; Navarro, F.; Pascual, Á.; Martínez-Martínez, L.; Oliver, A. Activity of Ceftazidime-Avibactam against Clinical and Isogenic Laboratory Pseudomonas aeruginosa Isolates Expressing Combinations of Most Relevant β-Lactam Resistance Mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6407–6410.
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62, e01379-18.
- Recio, R.; Villa, J.; Viedma, E.; Orellana, M.; Lora-Tamayo, J.; Chaves, F. Bacteraemia due to extensively drug-resistant Pseudomonas aeruginosa sequence type 235 high-risk clone: Facing the perfect storm. Int. J. Antimicrob. Agents 2018, 52, 172–179.
- Díaz-Cañestro, M.; Periañez, L.; Mulet, X.; Martin-Pena, M.L.; Fraile-Ribot, P.A.; Ayestarán, I.; Colomar, A.; Nuñez, B.; Maciá, M.; Novo, A.; et al. Ceftolozane/tazobactam for the treatment of multidrug resistant Pseudomonas aeruginosa: Experience from the Balearic Islands. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2191–2200.
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In Vivo Emergence of Resistance to Novel Cephalosporin–β-Lactamase Inhibitor Combinations through the Duplication of Amino Acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01117-17.
- Watson, M.E.; Burns, J.L.; Smith, A.L. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology 2004, 150, 2947–2958.
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science 2000, 288, 1251–1253.
- Hocquet, D.; Llanes, C.; Thouverez, M.; Kulasekara, H.D.; Bertrand, X.; Plésiat, P.; Mazel, D.; Miller, S.I. Evidence for induction of integron-based an-tibiotic resistance by the SOS response in a clinical setting. PLoS Pathog. 2012, 8, e1002778.
- Breidenstein, E.B.M.; Bains, M.; Hancock, R.E.W. Involvement of the Lon Protease in the SOS Response Triggered by Ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2012, 56, 2879–2887.
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 2020, 19, 331–342.
- Smith, P.A.; Romesberg, F.E. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat. Chem. Biol. 2007, 3, 549–556.
- Horii, T.; Ogawa, T.; Nakatani, T.; Hase, T.; Matsubara, H.; Ogawa, H. Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 1981, 27, 515–522.
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64.
- Sutton, M.D.; Smith, B.T.; Godoy, V.G.; Walker, G.C. The SOS response: Recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 2000, 34, 479–497.
- Gruber, A.J.; Erdem, A.L.; Sabat, G.; Karata, K.; Jaszczur, M.M.; Vo, D.D.; Olsen, T.M.; Woodgate, R.; Goodman, M.F.; Cox, M.M. A RecA Protein Surface Required for Activation of DNA Polymerase V. PLoS Genet. 2015, 11, e1005066.
- Naiman, K.; Pagès, V.; Fuchs, R.P. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res. 2016, 44, 7691–7699.
- Yakimov, A.; Bakhlanova, I.; Baitin, D. Targeting evolution of antibiotic resistance by SOS response inhibition. Comput. Struct. Biotechnol. J. 2021, 19, 777–783.
- Sniegowski, P.D.; Gerrish, P.J.; Lenski, R.E. Evolution of high mutation rates in experimental populations of E. coli. Nature 1997, 387, 703–705.
- Pomerantz, R.T.; Kurth, I.; Goodman, M.F.; O’Donnell, M.E. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat. Struct. Mol. Biol. 2013, 20, 748–755.
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8.
- Boshoff, H.I.M.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193.
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138.
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113.
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323.
- Taylor, P.K.; Yeung, A.T.; Hancock, R.E. Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies. J. Biotechnol. 2014, 191, 121–130.
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The Formation of Biofilms by Pseudomonas aeruginosa: A Review of the Natural and Synthetic Compounds Interfering with Control Mechanisms. Biomed Res. Int. 2015, 2015, 759348.
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199.
- Kang, D.; Turner, K.E.; Kirienko, N.V. PqsA Promotes Pyoverdine Production via Biofilm Formation. Pathogens 2017, 7, 3.
- Parkins, M.D.; Ceri, H.; Storey, D.G. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm for-mation. Mol. Microbiol. 2001, 40, 1215–1226.
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 544–553.
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273.
- Ha, D.-G.; O’Toole, G.A.; Ghannoum, M.; Parsek, M.; Whiteley, M.; Mukherjee, P.K. c-di-GMP and its Effects on Biofilm Formation and Dispersion: A Pseudomonas aeruginosa Review. Microbiol. Spectr. 2015, 3, MB-0003-2014.
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219.
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid Pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34.
- Mah, T.-F.; Pitts, B.; Pellock, B.; Walker, G.C.; Stewart, P.S.; O’Toole, G.A. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 2003, 426, 306–310.
- Zhang, L.; Mah, T.-F. Involvement of a Novel Efflux System in Biofilm-Specific Resistance to Antibiotics. J. Bacteriol. 2008, 190, 4447–4452.
- Zhang, L.; Hinz, A.J.; Nadeau, J.P.; Mah, T.F. Pseudomonas aeruginosa tssC1 links type VI secretion and biofilm-specific antibiotic resistance. J. Bacteriol. 2011, 193, 5510–5513.