Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Huntington’s disease (HD) is a debilitating neurodegenerative genetic disorder caused by an expanded polyglutamine-coding (CAG) trinucleotide repeat in the huntingtin (HTT) gene. HD behaves as a highly penetrant dominant disorder likely acting through a toxic gain of function by the mutant huntingtin protein. Widespread cellular degeneration of the medium spiny neurons of the caudate nucleus and putamen are responsible for the onset of symptomology that encompasses motor, cognitive, and behavioural abnormalities.
- Huntington’s disease
- pathogenesis
- therapeutics
1. Introduction
Huntington’s disease (HD), previously known as Huntington’s chorea, was succinctly and accurately characterised in 1872 by physician George Huntington. He described the occurrence of a unique form of chorea characterised by “its hereditary nature… tendency to insanity… its coming on, at least as a grave disease, only in adult life” [1]. Subsequently, researchers have described a characteristic degeneration of striatal medium spiny neurons of the basal ganglia which is responsible for the manifestation of symptoms that encompasses motor, cognitive, and behavioural abnormalities [2]. There are now well-established clinical tests and, to a lesser extent, biomarkers that can ascertain both symptom onset and severity over the disease course [3][4]. The genetic aetiology was defined as an expansion of a polyglutamine coding repeat in exon 1 of the huntingtin gene [5], which lead to the development of an accurate genetic test for diagnosis [6].
2. Molecular Mechanisms and Therapeutic Development
An assortment of mechanisms for HD pathogenesis have been proposed and investigated. Two core hypotheses constitute the rationale for research and therapeutic design: a gain of aberrant toxicity from mHTT expression and a dominant loss of normal activity from wild-type HTT as a result of mHTT expression. An overview of the current proposed pathogenic mechanisms and drug development based on these mechanistic pathways will be discussed (summarised in Figure 1 and Table 1).
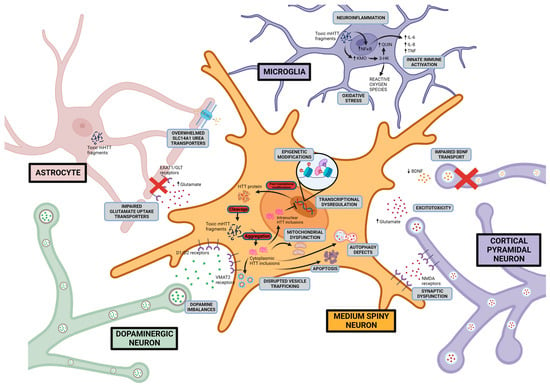
Figure 1. Proposed HD pathogenic mechanisms This figure shows possible Huntington disease mechanisms and the cellular location of the aberrant activity including excitotoxicity, dopamine imbalance, mitochondrial dysfunction, disruption of proteostasis, initiation of apoptotic pathways, transcriptional dysregulation, and neuroinflammation. Created with BioRender.com (accessed on 17 August 2023).
2.1. Excitotoxicity-Induced Neurodegeneration
Glutamate excitotoxicity as an initiator of neuronal death has been a longstanding hypothesis and research focus in neurodegenerative disorders [7][8][9][10]. In HD, it has been proposed that the neurodegeneration of the medium spiny neurons of the striatum is caused by excess glutamate in the synapses resulting from a lack of synaptic glutamate clearance and an increased glutamate release. An observed reduction in expression of astrocytic glutamate uptake receptors, GLT1, and GLAST has been reported in the striatum of HD mouse models and human post-mortem tissue, which may explain the elevated glutamate levels [11][12]. Excess synaptic glutamate has been proposed to lead to increased signalling of post-synaptic N-methyl-d-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and kainate receptors. Consequently, this may lead to an influx of calcium ions, chronic membrane depolarisation, oxidative stress, and activation of cell death pathways [7][8][9][10][13]. Evidence for NMDA receptor-mediated excitotoxicity was first shown through striatal injection of glutamate receptor agonists (glutamic acid, kainic acid, quinolinic acid) in rodents and non-human primates, resulting in medium spiny neuronal degeneration and HD motor dysfunction [14][15][16][17][18][19][20]. Further, studies investigating several HD mouse models provided evidence for an association between excessive NMDA receptor expression, receptor currents, and striatal degeneration [21][22][23]. Ionotropic glutamate receptors were also most pronounced in striatal medium spiny neurons compared to striatal interneurons, which may explain the preferential neurodegeneration of medium spiny neurons [24].
Treatments Targeting Excitotoxicity
NMDA receptor antagonists have been tested in clinical trials for their ability to reduce glutamate receptor overactivation. These compounds have included Memantine, an NMDA receptor antagonist that is FDA approved for the treatment of dementia symptoms in Alzheimer’s disease [25]. Memantine first showed efficacy in the 3-nitropropionic mouse model, where intraperitoneal injections of Memantine resulted in reduced striatal degeneration and improved motor and cognitive function [26]. A phase IV clinical trial was conducted involving 50 symptomatic HD patients that received either memantine or a placebo and were followed up with over 3 months. Unfortunately, the study showed no improvement to motor function between the treatment and control groups, although the researchers noted that longer studies are likely required to validate these findings [27]. Another NMDA receptor antagonist, Dimebon (laterepirdine), failed to showcase improvement in motor and cognitive function in a placebo-controlled phase III clinical trial involving 350 HD patients over the course of 6 months [28]. The NMDA receptor antagonist, amantadine was trialled in HD individuals for its efficacy in reducing parkinsonian dyskinesias [29][30]. The efficacy of amantadine in clinical trials has been mixed, with some trials reporting improvement to motor function and others indicating no significant differences [31][32][33].
Other treatments targeting excitotoxicity have been developed to limit the availability of extracellular synaptic glutamate. Excessive glutamate release may be diminished by blocking voltage-gated sodium channels. The sodium channel blocker Riluzole was investigated in HD patients for its beneficial effects in mediating excitotoxicity in mouse models [7]. However, a phase III placebo-controlled clinical trial conducted with 537 HD patients over a three-year period concluded that riluzole had no benefit for the treatment of HD, with no improvements to motor, cognitive, or behavioural function observed [34]. Another sodium channel blocker, BN82451, was shown to improve motor function, decrease atrophy, and extended survival in R6/2 HD mice [35]. However, a phase II placebo-controlled clinical trial involving 17 patients was terminated due to subject recruitment problems (www.clinicaltrials.gov, identifier NCT02231580, accessed on 17 August 2023).
Overall, NMDA antagonists and glutamate-lowering drugs have showed limited, sustained efficacy in the improvement of HD motor and cognitive deficiencies. However, several of these studies were underpowered, with a limited number of participants treated over a short period of time. The trials also recruited mainly symptomatic HD patients, for whom widespread degeneration of neurons would have already occurred. Assessment of the efficacy of treatments targeting excitotoxicity may be confounded by the consequences of widespread catastrophe brought upon by the cell death cascade reaching a point where recovery becomes unattainable. With evidence that suggests excitotoxicity may begin early in the disease phase [36][37][38], future clinical trials should consider whether administration of these therapeutics at an earlier disease stage may result in improved efficacy compared to what is currently observed.
2.2. Dopaminergic Imbalance
The dopaminergic pathway is thought to be dysfunctional in HD and responsible for movement-related symptoms including chorea, rigidity, and akinesia [39]. Studies examining the cerebrospinal fluid of HD patients have indicated an increase in dopamine levels during early stages of the disease, correlating to the onset of chorea [40]. Interestingly, dopamine levels decrease throughout disease course, and post-mortem HD brains show reduced levels of caudate and striatal dopamine correlating with rigidity and akinesia symptoms present during late stages of the disease [41]. PET studies have shown a significant decrease in dopamine availability in akinetic patients, resulting in reduced binding to striatal dopamine D1 and D2 receptors [42][43], dopamine transporter (DAT) [44], and vesicular monoamine transporter 2 (VMAT2) [45]. Findings in HD mouse models agree with human studies showing an initial increase in dopamine levels and dopamine release and binding to D1 and D2 receptors, followed by a progressive reduction with disease progression [46][47][48][49][50]. The exact mechanisms of dopamine regulation in the HD basal ganglia are unclear and a question remains as to whether dopamine alterations are a cause or consequence of the widespread neurodegeneration.
Treatments Targeting the Dopaminergic Pathway
Two FDA-approved drugs for the symptomatic treatment of HD-mediated chorea, tetrabenazine and deutetrabenazine, target the dopaminergic pathway through inhibition of the vesicular monoamine transporter (VMAT2 inhibitor), resulting in decreased bioavailability of dopamine in synapses and reduced dopamine signalling. Both tetrabenazine and deutetrabenazine ameliorated chorea and other motor symptoms in an HD mouse model [51]. These results were similarly replicated in human clinical trials showcasing improved motor function [52][53][54]. The deuterated form of tetrabenazine, deutetrabenazine improved on the half-life of tetrabenazine and allowed for administration at lower doses and reduced adverse effects [54][55]. Recently, another VMAT2 inhibitor, valbenazine, has shown success in a phase III clinical trial with preliminary results indicating a significant improvement in motor function (www.clinicaltrials.gov, identifier NCT04102579, accessed on 17 August 2023) [56].
Antipsychotic drugs are commonly prescribed off-label for HD chorea, psychiatric problems, sleep dysfunction, and weight loss despite no consistent evidence to support their therapeutic use [57]. Risperidone, generally used in the treatment of schizophrenia and bipolar disorders [58], is currently being assessed in a phase II clinical trial involving 12 participants (www.clinicaltrials.gov, identifier NCT04201834, accessed on 17 August 2023). A completed phase III clinical trial comparing tetrabenazine with two antipsychotics, olanzapine and tiapride, is expected to publish results (www.clinicaltrials.gov, identifier NCT00632645, accessed on 17 August 2023). Another antipsychotic, rolipram, was shown to be neuroprotective in an HD mouse model, and was hypothesised to act through its inhibition of phosphodiesterase and the second messenger dopamine cascade [59].
2.3. Mitochondrial Dysfunction
Evidence for mitochondrial dysfunction in HD stems from the observation that systemic administration of 3-nitropropionic acid, an irreversible inhibitor of complex II of the mitochondrial electron transport chain in both rodents and non-human primates, reproduced many of the behavioural and anatomical characteristics of HD [60][61]. Mutant huntingtin protein also appears to directly sequester the intracellular transport machinery and potentially interfere with both anterograde and retrograde trafficking of mitochondria to sites of high energy demand such as neuronal dendrites, axons, and synapses, culminating in overall synaptic degeneration and neurodegeneration [62][63][64]. As previously mentioned, metabolic energy deficits have been reported in HD patients, often shown as weight loss. Increased lactic acid and decreased glucose metabolism were observed in HD patients [65][66][67], and a reduction in circulating levels of branched chain amino acids typically involved in energy homeostasis was seen that is correlated with weight loss [68]. These changes were seen early in the disease before the onset of symptoms and suggest that metabolic deficits in HD may precede neuropathology and clinical symptoms [68].
In HD patient-derived cell lines and HD mouse models, a number of studies showcase abnormal ATP:ADP and phosphocreatine:inorganic phosphate (PCr:Pi) ratios, likely due to an impaired mitochondrial electron flow [69][70][71]. Isolated mitochondria from HD mouse models and lymphoblasts from HD patients showcased a depolarised mitochondrial membrane that became progressively disrupted with increasing polyglutamine repeat length [72][73]. A decrease in the activity of electron transport chain complex II (succinate dehydrogenase) and complex IV (cytochrome c oxidase) is suggested to explain the membrane depolarisation [74]. Further, it has been proposed that an altered electron flow through the compromised mitochondrial complexes may promote the formation of reactive oxygen species (ROS) including superoxide (O2•−), hydrogen peroxide (H2O2), hydroxyl radical (•OH), and peroxynitrite (ONOO−) [75], which leads to activation of downstream apoptotic mechanisms of cell death.
Treatments Targeting Mitochondrial Dysfunction
Assessment of therapeutics targeting HD mitochondrial dysfunction have been confounded by difficulties in optimal dosing, trial length, and accurate measurement of target engagement. Eicosapentaenoic acid, an omega-3 fatty acid used to treat hypertriglyceridemia, showcased improvements to motor and behavioural deficits in an HD mouse model [76]. However, a phase III placebo-controlled clinical trial involving 300 patients showed no improvement to UHDRS motor scores over 6 months with no alterations in neurodegeneration [77][78]. Similarly, coenzyme Q10 supplementation was found to be neuroprotective in an HD mouse model displaying improvements in motor deficits, delayed striatal atrophy, and prolonged lifespans [79]. However, two independent large placebo-controlled clinical trials showed no significant slowing of disease progression at all dosages studied [80], with one of the studies (2CARE) terminated early due to the occurrence of severe adverse effects [81]. Creatine, which contains antioxidant properties, delayed striatal degeneration and reduced mutant huntingtin protein aggregation in an HD mouse model [82]. Again, clinical trials were unsatisfactory and a phase III placebo-controlled clinical trial involving 553 patients followed up to 48 months showed no influence on motor and cognitive deficits [83][84]. The cytoprotective peptide SBT-20 acts to prevent lipid peroxidation and initiation of apoptosis. SBT-20 was effective in protecting against mutant huntingtin-induced mitochondrial and synaptic damage in cultured HD neurons [85]. However, no significant differences in mitochondrial function were seen between treatment and placebo groups in phase I/II clinical trials [86].
Some promising results have been reported for Triheptanoin, a triglyceride that catabolises into substrates for the Krebs cycle, targeting the amelioration of HD metabolic deficits. Following one month of treatment, the ratio between inorganic phosphate and phosphocreatine levels that is disrupted in HD [69][70][71] was remedied, along with improvement in motor deficits [87]. A phase II placebo-controlled study involving 100 participants followed over 12 months was recently completed, evaluating triheptanoin in pre-manifest HD individuals (www.clinicaltrials.gov, identifier NCT02453061, accessed on 17 August 2023). Resveratrol, an antifungal agent proposed to inhibit p53-mediated mitochondrial apoptotic oxidative stress is currently being assessed in a phase III placebo-controlled clinical trial involving 102 patients followed over 12 months (www.clinicaltrials.gov, identifier NCT02336633, accessed on 17 August 2023) [88]. The PPARα agonist Fenofibrate is also currently being evaluated in a phase II placebo-controlled clinical trial involving 20 participants followed over a period of 6 months for its ability to induce PGC-1α expression levels involved in mitochondrial fission and fusion (www.clinicaltrials.gov, identifier NCT03515213, accessed on 17 August 2023).
2.4. Neuroinflammation
Neuroinflammation in the scope of neurodegenerative disorders describes the activation of glial cells, notably astrocytes and microglia, which respond to neuronal damage and act to remove the dead cells and other constituents by phagocytosis. However, this beneficial role may be limited, as chronic neuroinflammation, as in the case of HD, results in the accumulation of soluble pro-inflammatory mediators including cytokines, prostaglandins, and nitric oxide that further exacerbate cellular degeneration [89]. Evidence for neuroinflammation in HD has been presented as the gradual accumulation of reactive microglia and reactive astrocytes in HD brain tissue throughout the disease course [90][91][92]. PET scans using 11C-(R)-PK11195 and 11C-PBR28, two tracers that are high-affinity ligands for the peripheral benzodiazepine receptor (PBR) and markers for glial activation, have revealed extensive glial neuroinflammation in the striatal and cortical regions of HD patients that correlated with disease progression [93][94][95][96][97][98].
Microglia undergo morphological changes and become activated upon exposure to inflammatory stimuli and switch from being a promoter of growth and neurogenesis to a motile phagocytic role [89][99][100]. Signatures of microglia activation including pro-inflammatory interleukin 1B, interleukin 6, interleukin 8, TNFα, and MMP have been detected in the HD striatum, globus pallidus, and cortical areas [101][102][103]. Moreover, it has been suggested that mutant huntingtin protein may activate microglia through NF-κB signalling [104][105][106] and the kyurenine pathway [107][108]. Astrocytes also undergo morphological changes (reactive astrogliosis), and studies have found increased numbers of both neurotoxic A1 [109] and neuroprotective A2 astrocytes in HD [110]. It is proposed that neurotoxic A1 reactive astrocytes may increase synaptic concentrations of proinflammatory cytokines and reactive oxygen species, decrease glutamate uptake, and contribute to membrane depolarisation through impaired potassium homeostasis [109][111][112]. Conversely, neuroprotective A2 type astrocytes produce antioxidants and initiate reconstruction of damaged neuronal circuits [113][114].
Treatments Targeting Neuroinflammation
The small molecule compound Neflamapimod was of interest for its ability to inhibit proinflammatory kinases including tumour necrosis factor and interleukin-1β and reduce the number of reactive microglia [115][116]. Neflamapimod has been shown to reduce synaptic dysfunction and slow disease progression in Alzheimer’s disease patients [115][116]. Unfortunately, a phase II clinical trial comparing Neflamapimod against a placebo in 15 HD patients was prematurely terminated due to the COVID-19 pandemic (www.clinicaltrials.gov, identifier NCT03980938, accessed on 17 August 2023). The antibody VX15/2503 (pepinemab) raised against the transmembrane signalling molecule SEMA4D, which plays a role in regulating the activation of reactive microglia and astrocytes [117], was investigated in a placebo-controlled phase II clinical trial involving 256 patients followed over a period of 18 months [117][118]. Results indicated VX15/2503 resulted in a retainment of brain volume compared to the placebo group and showed improvement in cognitive measures, despite not meeting its primary endpoint [117][118]. Laquinimod, a quinoline-3-carboxamide derivate with anti-inflammatory properties, downregulates expression of the pro-apoptotic molecule Bax and reduces caspase-initiated apoptotic pathways [119]. Treatment with Laquinimod reduced striatal and cortical atrophy and improved motor function in an HD mouse model [120][121]. However, a phase II placebo-controlled clinical trial with Laquinimod in 15 patients over a follow up period of 12 months showed no significant change in 11C-PBR28 neuroinflammation marker levels between treatment and placebo groups [122]. The antibiotic minocycline was assessed for its anti-apoptotic effects and inhibition of reactive microgliosis [123][124] in a phase III placebo-controlled clinical trial involving 114 patients followed over 18 months (IND 60943). Unfortunately, minocycline did not achieve a >25% improvement in total functional capacity (measure of HD progression) and therefore was not continued in further trials [125].
2.5. mHTT Protein Misfolding and Endoplasmic Reticulum Stress
Mutant huntingtin protein remains an important pathogenic centerpiece that links neurodegeneration to abnormal protein degradation, endoplasmic reticulum (ER) stress, disrupted cellular trafficking, initiation of apoptotic pathways, and synaptic dysfunction. Toxic mutant huntingtin oligomer species interfere with ER-associated degradation components (ERAD) resulting in ER stress and the activation of the unfolded protein response [126][127]. The inhibition of ERAD leading to the accumulation of unfolded mutant huntingtin proteins and ER stress was observed in HD mouse models and post-mortem HD patients [128][129][130][131].
Lysosome-mediated macroautophagy plays a key role in the removal of misfolded proteins including mutant huntingtin [132]. Mutant huntingtin has been shown to stimulate the formation of autophagosomes through the inactivation of the mTOR kinase pathway [133][134]. However, despite the increase in autophagosome formation, cargo loading into autophagosomes is impaired in HD [134][135]. Therefore, despite an increase in the number of autophagic vesicles, mutant huntingtin and damaged organelles are not being degraded and instead are left to accumulate in the cytoplasm, where they continue to be toxic [136].
Treatments Directly Targeting mHTT Protein
Inhibition of the mTOR pathway induces mutant huntingtin protein autophagy and has demonstrated neuroprotective effects in HD cell and animal models. Rapamycin, an inhibitor of mTOR, reduced mutant huntingtin aggregates and neuronal atrophy in HD fly and mouse models [133]. Rapamycin has also been combined with lithium as a possible treatment option. Lithium induces autophagy through an mTOR-independent mechanism by inhibiting inositol monophosphatase. This dual approach seems to produce additive effects in an HD fly model [137]. Another antagonist to mTOR, metformin, reduced mutant huntingtin protein aggregation and reduced early behavioural defects in an HD mouse model [138]. In HD patients, metformin was associated with an improvement on cognitive tests but did not show a significant improvement in motor function [139].
The neuroprotective sigma-1 receptor is a molecular chaperone that is activated following ER stress [140][141]. Pridopidine, a sigma-1 receptor agonist was shown to increase brain-derived neurotrophic factor (BDNF) levels, reduce mutant huntingtin protein aggregation, and reduce motor impairments in an HD mouse model [142]. Early clinical trials suggested significant improvement to motor function [143][144][145]; however, a placebo-control involving 397 HD patients followed over a period of 26 weeks showed no improvement in UHDRS motor scores and the results of previous trials were not replicated [146].
Other studies have focused on stabilising the mutant huntingtin protein, thereby reducing its toxicity and propensity to form aggregates. Through screening of a library of natural compounds, Wanker and colleagues identified Epigallocatechin-3-gallate (EGCG), a catechin commonly found in green tea, which reduced mutant huntingtin misfolding and aggregation in vitro and decreased neurodegeneration in an HD fly model [147]. A completed phase II placebo-controlled clinical trial involving 54 patients followed over 12 months is expected to publish results (www.clinicaltrials.gov, identifier NCT01357681, accessed on 17 August 2023). Increased transglutaminase activity is thought to contribute to the misfolding of mutant huntingtin and formation of aggregates [148]. Cystamine, a competitive inhibitor of transglutaminase demonstrated improved motor function and overall survival in an HD mouse model, although the quantity and size of aggregates remained unchanged [149]. Unfortunately, a phase II/III placebo-controlled trial investigating cysteamine, the reduced form of Cystamine, in 96 HD patients followed over a period of 18 months showed no statistically significant improvement motor function [150]. Mutant huntingtin aggregation may be due to covalent attractions with metal ions, copper, and iron. Metal chelators including the 8-hydroxyquinoline analog named PBT2 showcased efficacy in an HD mouse model with improvements to motor deficits and extended lifespans [151]. A completed phase II placebo-controlled clinical trial involving 109 patients over a period of 26 weeks established safe and effective dose ranges for further evaluation in larger trials [152].
3. Invasive Therapeutics
Deep brain stimulation (DBS) for HD is primarily targeted at the globus pallidus and provides symptomatic relief for chorea. The exact mechanism of DBS is not well understood; however, studies suggest DBS alters electrical and neurochemical activity of neurons near the input electrode, which may confer a local or network-wide effect [153]. An initial pilot study with three individuals showed a reduction in chorea and improvement in quality-of-life scores but no significant effect on dystonia [154]. A phase II clinical trial was recently completed that examined the efficacy of DBS in 48 HD patients (www.clinicaltrials.gov, identifier NCT02535884, accessed on 17 August 2023). Another invasive technique, extracranial stereotactic radioablation of the thalamic region, has shown effectiveness in alleviating tremor symptoms present in Parkinson’s disease [155][156]. A clinical trial is currently evaluating the safety and effectiveness of extracranial stereotactic radioablation of the thalamus or globus pallidus in HD patients (www.clinicaltrials.gov, identifier NCT02252380, accessed on 17 August 2023).
Stem cell transplant therapies aim to replace damaged neurons in affected HD brain areas with new neurons derived from stem cells. Sources of stem cells include mesenchymal stem cells from bone marrow [157] or adipose tissue [158], embryonic stem cells from in vitro fertilized eggs [159], neural stem cells from brain tissue [160], or induced pluripotent stem cells [161]. The safety and efficacy of bone marrow-derived stem cells was evaluated in a phase I/II clinical trial involving 50 participants (www.clinicaltrials.gov, identifier NCT01834053, accessed on 17 August 2023). Results from the trial have not yet been released. An ongoing phase I/II dose escalation trial is investigating the effects of three intravenous injections of stem cells spread across 3 months. Individuals will be monitored for up to five years for safety and efficacy (www.clinicaltrials.gov, identifiers NCT03252535, NCT02728115, accessed on 17 August 2023). Another ongoing clinical trial is evaluating the efficacy of autologous stem cell isolates from adipose tissue in multiple neuropathies including HD (www.clinicaltrials.gov, identifier NCT03297177, accessed on 17 August 2023).
4. Genetic Mechanisms and Therapeutics
Huntingtin and mutant huntingtin interact with a wide variety of transcription factors, chromatin remodelling proteins, basal transcriptional machinery, and non-coding RNAs. Both wild-type huntingtin and mutant huntingtin can directly bind to DNA at promoter sequences or intronic/intergenic regions to alter gene expression [162][163]. Transcriptional dysregulation is not restricted to areas of the brain, but rather the ubiquitous expression of HTT alters transcript profiles across non-neuronal tissues such as blood [164], muscle [165][166], and liver [167]. Gene expression studies including microarray profiling [168][169], RNA-seq [170], and, recently, single cell RNA-seq [171][172][173] studies have identified several differentially expressed genes between HD and controls in post-mortem brain, HD patient cell lines, and HD animal models. Nevertheless, it is important to note that not all changes in gene expression in HD play a role in pathogenicity and that they may instead reflect the activation of compensatory mechanisms to offset toxic effects [174][175].
4.1. BDNF
The brain-derived neurotrophic factor (BDNF) gene encodes a small nerve growth factor (neurotrophin) protein that promotes neuronal growth, maturation, survival, and synaptic activity [176][177]. Loss of BDNF protein is well documented in post-mortem HD brains and can lead to impaired synaptic transmission and loss of neuronal plasticity and contribute to medium spiny neuronal death [178][179]. Depletion of cortical BDNF levels have been shown to exacerbate HD symptoms in mouse models [179]. In contrast, overexpression of BDNF rescued striatal atrophy and ameliorated motor dysfunction in the same mouse models [180]. The loss in BDNF was proposed to be due to a loss of function of wild-type huntingtin protein. Wild-type huntingtin was observed to promote the expression of BDNF and regulate its transport to synapses [176][181]. BDNF and its associated trophic pathways have been key targets for therapeutic development [182].
4.2. CREB/CBP
Numerous reports have described transcriptional dysregulation of the CREB (cAMP response element binding protein) pathway in HD [183][184]. CREB belongs to a family of basic leucine zipper transcription factors that play a role in a wide range of physiological processes that include neuronal plasticity, long term memory function, neuronal differentiation, and survival [185]. Additionally, coactivators of CREB, notably CREB-binding protein (CBP) and p300, play important roles with its acetyltransferase activity and ability to recruit and initiate transcription factors such as p53, c-Jun, c-myc, E2F, and the RNA polymerase II transcriptional machinery [162]. In HD, dysregulation of CREB-mediated transcription and/or activity of its cofactors causes a reduction in BDNF levels [186], altered NMDA signalling, and diminished activation of the MAPK/ERK and protein kinase A pathways [185][187]. Importantly CBP has been shown to sequester with mutant huntingtin protein in nuclear inclusions, which may in turn reduce CREB transcriptional activity. This sequestration was hypothesised to increase striatal cell vulnerability in HD mouse models [183][188].
4.3. REST
RE1 silencing transcription factor (REST) (also known as neuron-restrictive silencer factor, NRSF) is a Krüppel-type zinc finger protein that acts as a transcriptional repressor of neuronal genes in non-neuronal cells and is important for cell fate and the maintenance of cell identity [189]. In HD, REST has been shown to translocate to the nucleus, where it represses cortical transcripts including BDNF and a variety of HD-related non-coding RNAs (miR-9, miR-124) [176][190]. Wild-type huntingtin protein has been shown to inhibit REST activity by a sequestration mechanism, resulting in the retainment of REST in the cytoplasm. This mechanism is reduced with mutant huntingtin and results in increased translocation of REST into the nucleus and repression of its downstream effectors [190].
4.4. PGC1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is a transcriptional co-activator that regulates mitochondrial biogenesis, energy homeostasis, and antioxidant defences [191]. Involvement of PGC-1α in HD was first suggested by the observation that PGC-1α knockout mice exhibit mitochondrial dysfunction, striatal neurodegeneration, and motor deficits resembling HD [192][193]. Polymorphisms in PGC-1α have been shown to modify the age of HD onset [194][195], and decreased expression of PGC-1α was observed in post-mortem brains of HD patients and in HD mouse models [196][197]. Further, striatal lentiviral administration of PGC-1α in an HD mouse model ameliorated neurodegeneration and mutant huntingtin aggregation [196]. The decrease in PGC-1α expression was proposed to be due to the effects of mutant huntingtin, which interferes with the CREB/TAF4-mediated activation of PGC-1α by inhibiting the ability of CREB/TAF4 to bind to the PGC-1α promoter.
DNA Targeting Approaches
DNA approaches directly target the HTT DNA sequence, altering its transcriptional activity or completely removing the deleterious mutation by genomic editing. Zinc finger proteins (ZFP) contain a DNA-binding motif that can be modified to bind to certain regions of interest. ZFP are often fused to transcriptional activators/repressors to alter target gene expression or fused to a nuclease (usually FokI) to cleave at specific sites for precise gene editing [198]. Zinc finger transcriptional repressors targeting the expanded CAG repeat have shown success in patient-derived fibroblasts and stem cell-derived human neurons with up to 90% repression of mHTT and less than 15% repression of the wild-type HTT [199]. Experiments in HD mouse models also demonstrate efficacy, exhibiting similar levels of repression, and were able to ameliorate several cognitive and motor deficits [199]. Transcription activator-like effectors (TALE) are other DNA-targeting agents comprised of a TAL effector DNA binding domain that can be engineered to bind specific locations. Similarly to ZFP, TALE are often fused to either a transcriptional activator/repressor or nucleases (usually FokI) [200]. TALE fused to a transcriptional repressor designed against mHTT allele specific SNPs (rs762855, rs3856973, rs2024115) was able to achieve allele selectivity in human HD fibroblasts [201].
The CRISPR-Cas9 system has been exploited to cleave DNA at a specific location of choice mediated by a synthetic guide RNA (gRNA) apparatus. An mHTT allele selective CRISPR-Cas9 approach utilising dual gRNAs specific for mHTT SNPs was successful in removal of the promotor region and transcription start site and the polyglutamine expansion of the HTT gene and produced permanent inactivation of the mHTT allele in patient-derived fibroblasts [202]. Further, CRISPR-Cas9-catalysed selective excision of the mHTT allele in Q140 HD knock-in mice attenuated early neuropathology and motor deficits and depleted the formation of aggregates; however, surprisingly, overall survival was not affected [203].
RNA-Targeting Approaches
An alternative approach is to target the HTT transcript. RNA interference (RNAi) utilizes the natural defence system against invading pathogenic RNA molecules to ‘silence’ a gene of interest. Double-stranded RNA (including small interfering RNA, siRNA; short hairpin RNA, shRNA; and microRNA, miRNA) complementary to the transcript of interest can be synthesised and introduced into the cell where it binds to the mRNA and targets it for degradation by argonaute 2 [204][205]. In HD, double stranded RNA has been designed to bind the HTT transcript (non-selective) or exclusively to the mHTT allele via the expanded CAG repeat or regions with allele-specific polymorphisms or small insertions and deletions (selective). Intrastriatal injection of adeno-associated virus (AAV)-containing non-selective HTT-targeting siRNA, shRNA, or miRNA into HD mouse models showed efficacy in ameliorating HD phenotypes [206][207][208][209]. A phase I/II proof of concept trial is currently recruiting HD patients to evaluate AAV-mediated delivery of a non-selective HTT-targeting miRNA (AMT-130) (www.clinicaltrials.gov, identifier NCT04120493, accessed on 17 August 2023) [210][211].
Antisense oligonucleotides (ASO) are 8–50 nucleotide-long single-stranded DNA molecules synthesised to bind existing pre-mRNA of a targeted gene and mark it for degradation by ribonuclease H [204][212]. The success of ASO in the treatment of genetic diseases has been shown in two FDA approved therapies: Nusinersen for the treatment of spinal muscular atrophy [213] and eteplirsen for the treatment of Duchenne’s muscular dystrophy [214]. In HD, a non-selective ASO termed RG6042 (previously IONIS-HTTRX) demonstrated efficacy in three different HD mouse models (YAC128, R6/2, BACHD) with approximately 80% reduction of mHTT mRNA and 63% reduction of mutant huntingtin protein. Reversal of the disease phenotype was also documented in all three HD mouse models [215]. These results were further replicated in a large mammalian primate model, which likewise demonstrated 50% reduction of mHTT mRNA in the cortex and 20% reduction in the striatum [215]. Following these promising results, a phase I/II dose escalation trial was carried out with forty-six individuals receiving either intrathecal injections of RG6042 ASO or placebo over a four month period [216]. A 40% reduction in mHTT in the CSF was observed that was dose dependent, possibly reflecting a 55–85% reduction of cortical mutant huntingtin protein and 20–50% reduction of striatal mutant huntingtin protein [216]. A subsequent phase III trial was undertaken with 791 HD individuals placed into 3 groups, with intrathecal injection of RG6042 every 8 weeks, intrathecal injection of RG6042 every 16 weeks, or placebo. Unfortunately, RG6042 was found to exacerbate disease symptoms in the group receiving RG6042 every 8 weeks, with consistently worse scores on UHDRS and total functional capacity compared to the placebo. The individuals given RG6042 every 16 weeks showed no significant differences from the placebo. With the results suggesting that RG6042 was not benefiting patients, dosing was discontinued in March 2021 (www.clinicaltrials.gov, identifier NCT03761849, accessed on 17 August 2023). A dose-finding phase II clinical trial was subsequently initiated to evaluate the safety and efficacy of RG6042 in younger patients with a lower disease burden determined by the CAG-repeat age product score [217].
Allele-specific treatments aim to avoid the complications derived from a reduction in wild-type HTT. Two clinical trials have been completed investigating ASO-targeting HD-associated SNP rs362307 (www.clinicaltrials.gov, identifier NCT03225833, accessed on 17 August 2023) and ASO-targeting HD-associated SNP rs362331 (www.clinicaltrials.gov, identifier NCT03225846, accessed on 17 August 2023). However, neither ASO was associated with a significant decrease in mutant huntingtin protein levels in patients’ cerebrospinal fluid compared to the placebo group.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241613021
References
- Huntington, G. On chorea. 1872. Med. Surg. Rep. 1872, 26, 317–321.
- Nance, M.A. Huntington Disease: Clinical, Genetic, and Social Aspects. J. Geriatr. Psychiatry Neurol. 1998, 11, 61–70.
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216.
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005.
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983.
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 219–226.
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221.
- Salinska, E.; Danysz, W.; Lazarewicz, J.W. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005, 43, 322–339.
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387.
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18.
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067.
- Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485.
- Sepers, M.D.; Raymond, L.A. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 2014, 19, 990–996.
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659.
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171.
- Bordelon, Y.M.; Chesselet, M.-F.; Nelson, D.; Welsh, F.; Erecińska, M. Energetic Dysfunction in Quinolinic Acid-Lesioned Rat Striatum. J. Neurochem. 1997, 69, 1629–1639.
- Emerich, D.F.; Thanos, C.G.; Goddard, M.; Skinner, S.J.; Geany, M.S.; Bell, W.J.; Bintz, B.; Schneider, P.; Chu, Y.; Babu, R.S.; et al. Extensive neuroprotection by choroid plexus transplants in excitotoxin lesioned monkeys. Neurobiol. Dis. 2006, 23, 471–480.
- Foster, A.C.; Miller, L.P.; Oldendorf, W.H.; Schwarcz, R. Studies on the disposition of quinolinic acid after intracerebral or systemic administration in the rat. Exp. Neurol. 1984, 84, 428–440.
- Coyle, J.T.; Schwarcz, R. Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 1976, 263, 244–246.
- McGeer, E.G.; McGeer, P.L. Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature 1976, 263, 517–519.
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190.
- Joshi, P.R.; Wu, N.P.; Andre, V.M.; Cummings, D.M.; Cepeda, C.; Joyce, J.A.; Carroll, J.B.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S.; et al. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 2009, 29, 2414–2427.
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 2009, 29, 3200–3205.
- Rigby, M.; Le Bourdelles, B.; Heavens, R.P.; Kelly, S.; Smith, D.; Butler, A.; Hammans, R.; Hills, R.; Xuereb, J.H.; Hill, R.G.; et al. The messenger RNAs for the N-methyl-D-aspartate receptor subunits show region-specific expression of different subunit composition in the human brain. Neuroscience 1996, 73, 429–447.
- van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247.
- Lee, S.-T.; Chu, K.; Park, J.-E.; Kang, L.; Ko, S.-Y.; Jung, K.-H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207.
- Ondo, W.G.; Mejia, N.I.; Hunter, C.B. A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Park. Relat. Disord. 2007, 13, 453–454.
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol. 2010, 67, 154–160.
- Crosby, N.J.; Deane, K.H.; Clarke, C.E. Amantadine for dyskinesia in Parkinson’s disease. Cochrane Database Syst. Rev. 2003, 2003, CD003467.
- Luginger, E.; Wenning, G.K.; Bosch, S.; Poewe, W. Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2000, 15, 873–878.
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002, 59, 694–699.
- Lucetti, C.; Del Dotto, P.; Gambaccini, G.; Dell’ Agnello, G.; Bernardini, S.; Rossi, G.; Murri, L.; Bonuccelli, U. IV amantadine improves chorea in Huntington’s disease: An acute randomized, controlled study. Neurology 2003, 60, 1995–1997.
- O’Suilleabhain, P.; Dewey, J.; Richard, B. A Randomized Trial of Amantadine in Huntington Disease. Arch. Neurol. 2003, 60, 996–998.
- Landwehrmeyer, G.B.; Dubois, B.; de Yebenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272.
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.E.; Beal, M.F. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2003, 86, 267–272.
- Levine, M.S.; Klapstein, G.J.; Koppel, A.; Gruen, E.; Cepeda, C.; Vargas, M.E.; Jokel, E.S.; Carpenter, E.M.; Zanjani, H.; Hurst, R.S.; et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 1999, 58, 515–532.
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernandez, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 2001, 66, 525–539.
- Starling, A.J.; Andre, V.M.; Cepeda, C.; de Lima, M.; Chandler, S.H.; Levine, M.S. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington’s disease. J. Neurosci. Res. 2005, 82, 377–386.
- Cepeda, C.; Murphy, K.P.; Parent, M.; Levine, M.S. The role of dopamine in Huntington’s disease. Prog. Brain Res. 2014, 211, 235–254.
- Garrett, M.C.; Soares-da-Silva, P. Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in Huntington’s disease: Evidence for an overactive dopaminergic brain transmission. J. Neurochem. 1992, 58, 101–106.
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 1987, 22, 386–389.
- Richfield, E.K.; O’Brien, C.F.; Eskin, T.; Shoulson, I. Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci. Lett. 1991, 132, 121–126.
- van Oostrom, J.C.; Dekker, M.; Willemsen, A.T.; de Jong, B.M.; Roos, R.A.; Leenders, K.L. Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington’s disease. Eur. J. Neurol. 2009, 16, 226–231.
- Ginovart, N.; Lundin, A.; Farde, L.; Halldin, C.; Backman, L.; Swahn, C.G.; Pauli, S.; Sedvall, G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain 1997, 120 Pt 3, 503–514.
- Bohnen, N.I.; Koeppe, R.A.; Meyer, P.; Ficaro, E.; Wernette, K.; Kilbourn, M.R.; Kuhl, D.E.; Frey, K.A.; Albin, R.L. Decreased striatal monoaminergic terminals in Huntington disease. Neurology 2000, 54, 1753–1759.
- Hickey, M.A.; Reynolds, G.P.; Morton, A.J. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington’s disease. J. Neurochem. 2002, 81, 46–59.
- Johnson, M.A.; Rajan, V.; Miller, C.E.; Wightman, R.M. Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2006, 97, 737–746.
- Callahan, J.W.; Abercrombie, E.D. In vivo Dopamine Efflux is Decreased in Striatum of both Fragment (R6/2) and Full-Length (YAC128) Transgenic Mouse Models of Huntington’s Disease. Front. Syst. Neurosci. 2011, 5, 61.
- Ortiz, A.N.; Kurth, B.J.; Osterhaus, G.L.; Johnson, M.A. Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J Neurochem. 2010, 112, 755–761.
- Koch, E.T.; Raymond, L.A. Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 2019, 97, 1636–1654.
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol. Neurodegener. 2010, 5, 18.
- Kegelmeyer, D.A.; Kloos, A.D.; Fritz, N.E.; Fiumedora, M.M.; White, S.E.; Kostyk, S.K. Impact of tetrabenazine on gait and functional mobility in individuals with Huntington’s disease. J. Neurol. Sci. 2014, 347, 219–223.
- Tang, T.S.; Chen, X.; Liu, J.; Bezprozvanny, I. Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J. Neurosci. 2007, 27, 7899–7910.
- Frank, S.; Testa, C.M.; Stamler, D.; Kayson, E.; Davis, C.; Edmondson, M.C.; Kinel, S.; Leavitt, B.; Oakes, D.; O’neill, C. Effect of deutetrabenazine on chorea among patients with Huntington disease: A randomized clinical trial. Jama 2016, 316, 40–50.
- Reilmann, R. Deutetrabenazine-Not a Revolution but Welcome Evolution for Treating Chorea in Huntington Disease. JAMA Neurol. 2016, 73, 1404–1406.
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Huntington Study Group, K.-H.D.C. Safety and efficacy of valbenazine for the treatment of chorea associated with Huntington’s disease (KINECT-HD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2023, 22, 494–504.
- Unti, E.; Mazzucchi, S.; Palermo, G.; Bonuccelli, U.; Ceravolo, R. Antipsychotic drugs in Huntington’s disease. Expert Rev. Neurother. 2017, 17, 227–237.
- Brown, C.S.; Markowitz, J.S.; Moore, T.R.; Parker, N.G. Atypical antipsychotics: Part II: Adverse effects, drug interactions, and costs. Ann. Pharmacother. 1999, 33, 210–217.
- DeMarch, Z.; Giampa, C.; Patassini, S.; Bernardi, G.; Fusco, F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2008, 30, 375–387.
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192.
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109.
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420.
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209.
- Chang, D.T.; Rintoul, G.L.; Pandipati, S.; Reynolds, I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006, 22, 388–400.
- Antonini, A.; Leenders, K.L.; Spiegel, R.; Meier, D.; Vontobel, P.; Weigell-Weber, M.; Sanchez-Pernaute, R.; de Yebenez, J.G.; Boesiger, P.; Weindl, A.; et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain 1996, 119 Pt 6, 2085–2095.
- Mazziotta, J.C.; Phelps, M.E.; Pahl, J.J.; Huang, S.C.; Baxter, L.R.; Riege, W.H.; Hoffman, J.M.; Kuhl, D.E.; Lanto, A.B.; Wapenski, J.A.; et al. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington’s disease. N. Engl. J. Med. 1987, 316, 357–362.
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 1993, 43, 2689–2695.
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647.
- Mochel, F.; N’Guyen, T.-M.; Deelchand, D.; Rinaldi, D.; Valabregue, R.; Wary, C.; Carlier, P.G.; Durr, A.; Henry, P.-G. Abnormal response to cortical activation in early stages of Huntington disease. Mov. Disord. 2012, 27, 907–910.
- Mochel, F.; Durant, B.; Meng, X.; O’Callaghan, J.; Yu, H.; Brouillet, E.; Wheeler, V.C.; Humbert, S.; Schiffmann, R.; Durr, A. Early alterations of brain cellular energy homeostasis in Huntington disease models. J. Biol. Chem. 2012, 287, 1361–1370.
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.; Gusella, J.F.; Laramie, J.M.; Myers, R.H.; Lesort, M.; et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880.
- Milakovic, T.; Quintanilla, R.A.; Johnson, G.V. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: Functional consequences. J. Biol. Chem. 2006, 281, 34785–34795.
- Sawa, A.; Wiegand, G.W.; Cooper, J.; Margolis, R.L.; Sharp, A.H.; Lawler, J.F., Jr.; Greenamyre, J.T.; Snyder, S.H.; Ross, C.A. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999, 5, 1194–1198.
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer International Publishing: Cham, Swizerland, 2018; pp. 59–83.
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144.
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Lu, G.; Barakauskas, V.E.; Barr, A.M.; Honer, W.G.; Hayden, M.R.; Leavitt, B.R. Ethyl-EPA treatment improves motor dysfunction, but not neurodegeneration in the YAC128 mouse model of Huntington disease. Exp. Neurol. 2005, 196, 266–272.
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589.
- Ferreira, J.J.; Rosser, A.; Craufurd, D.; Squitieri, F.; Mallard, N.; Landwehrmeyer, B. Ethyl-eicosapentaenoic acid treatment in Huntington’s disease: A placebo-controlled clinical trial. Mov. Disord. 2015, 30, 1426–1429.
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599.
- Huntington Study, G. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 2001, 57, 397–404.
- Annoucement of 2CARE Early Study Closure. 2014. Available online: http://hdsa.org/wp-content/uploads/2015/01/Announcement-of-2CARE-Early-Study-Closure.pdf (accessed on 17 August 2023).
- Ferrante, R.J.; Andreassen, O.A.; Jenkins, B.G.; Dedeoglu, A.; Kuemmerle, S.; Kubilus, J.K.; Kaddurah-Daouk, R.; Hersch, S.M.; Beal, M.F. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J. Neurosci. 2000, 20, 4389–4397.
- Hersch, S.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′ dG. Neurology 2006, 66, 250–252.
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.L.; Meyers, C.M.; Nahin, R.; Rosas, H.D.; Huntington Study Group, C.-E.I. Coordinators The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology 2017, 89, 594–601.
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753.
- van Diemen, M.P.J.; Hart, E.P.; Abbruscato, A.; Mead, L.; van Beelen, I.; Bergheanu, S.C.; Hameeteman, P.W.; Coppen, E.; Winder, J.Y.; Moerland, M.; et al. Safety, pharmacokinetics and pharmacodynamics of SBT-020 in patients with early stage Huntington’s disease, a 2-part study. Br. J. Clin. Pharmacol. 2021, 87, 2290–2302.
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495.
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169.
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373.
- Kassubek, J.; Juengling, F.D.; Kioschies, T.; Henkel, K.; Karitzky, J.; Kramer, B.; Ecker, D.; Andrich, J.; Saft, C.; Kraus, P.; et al. Topography of cerebral atrophy in early Huntington’s disease: A voxel based morphometric MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 213–220.
- Hobbs, N.Z.; Barnes, J.; Frost, C.; Henley, S.M.; Wild, E.J.; Macdonald, K.; Barker, R.A.; Scahill, R.I.; Fox, N.C.; Tabrizi, S.J. Onset and progression of pathologic atrophy in Huntington disease: A longitudinal MR imaging study. AJNR Am. J. Neuroradiol. 2010, 31, 1036–1041.
- Pagano, G.; Niccolini, F.; Politis, M. Current status of PET imaging in Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1171–1182.
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643.
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007, 72, 148–151.
- Politis, M.; Pavese, N.; Tai, Y.F.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Hypothalamic involvement in Huntington’s disease: An in vivo PET study. Brain 2008, 131, 2860–2869.
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of -(R)-PK 11195 and PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage 2010, 49, 2924–2932.
- Collste, K.; Forsberg, A.; Varrone, A.; Amini, N.; Aeinehband, S.; Yakushev, I.; Halldin, C.; Farde, L.; Cervenka, S. Test-retest reproducibility of PBR28 binding to TSPO in healthy control subjects. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 173–183.
- Datta, G.; Colasanti, A.; Kalk, N.; Owen, D.; Scott, G.; Rabiner, E.A.; Gunn, R.N.; Lingford-Hughes, A.; Malik, O.; Ciccarelli, O.; et al. (11)C-PBR28 and (18)F-PBR111 Detect White Matter Inflammatory Heterogeneity in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1477–1482.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat Neurosci. 2018, 21, 1359–1369.
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258.
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172.
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 2017, 9, 193.
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008, 205, 1869–1877.
- Kraft, A.D.; Kaltenbach, L.S.; Lo, D.C.; Harry, G.J. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging 2012, 33, 621.e17–621.e33.
- Khoshnan, A.; Ko, J.; Watkin, E.E.; Paige, L.A.; Reinhart, P.H.; Patterson, P.H. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 2004, 24, 7999–8008.
- Khoshnan, A.; Patterson, P.H. The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol. Dis. 2011, 43, 305–311.
- Schwarcz, R.; Guidetti, P.; Sathyasaikumar, K.V.; Muchowski, P.J. Of mice, rats and men: Revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog. Neurobiol. 2010, 90, 230–245.
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531.
- Liddelow, S.A. and Barres, B. A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967.
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546.
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87.
- Guttenplan, K.A.; Liddelow, S.A. Play It Again, SAM: Macrophages Control Peripheral Fat Metabolism. Trends Immunol. 2018, 39, 81–82.
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410.
- Robel, S.; Berninger, B.; Gotz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104.
- Alam, J.; Blackburn, K.; Patrick, D. Neflamapimod: Clinical Phase 2b-Ready Oral Small Molecule Inhibitor of p38alpha to Reverse Synaptic Dysfunction in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2017, 4, 273–278.
- Prins, N.D.; Harrison, J.E.; Chu, H.-M.; Blackburn, K.; Alam, J.J.; Scheltens, P.; Arnold; Coskinas; Gonzales; Joseph; et al. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 106.
- Feigin, A.; Evans, E.E.; Fisher, T.L.; Leonard, J.E.; Smith, E.S.; Reader, A.; Mishra, V.; Manber, R.; Walters, K.A.; Kowarski, L.; et al. Pepinemab antibody blockade of SEMA4D in early Huntington’s disease: A randomized, placebo-controlled, phase 2 trial. Nat. Med. 2022, 28, 2183–2193.
- Fernandes, J. Treatment with VX15 may protect brain in early phase Huntington’s, data shows. Huntington’s Disease News. 2017. Available online: https://huntingtonsdiseasenews.com/news/vx15-protect-brain-early-phase-huntingtons/ (accessed on 17 August 2023).
- Ehrnhoefer, D.E.; Caron, N.S.; Deng, Y.; Qiu, X.; Tsang, M.; Hayden, M.R. Laquinimod decreases Bax expression and reduces caspase-6 activation in neurons. Exp. Neurol. 2016, 283, 121–128.
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016, 6, 31652.
- Garcia-Miralles, M.; Yusof, N.; Tan, J.Y.; Radulescu, C.I.; Sidik, H.; Tan, L.J.; Belinson, H.; Zach, N.; Hayden, M.R.; Pouladi, M.A. Laquinimod Treatment Improves Myelination Deficits at the Transcriptional and Ultrastructural Levels in the YAC128 Mouse Model of Huntington Disease. Mol. Neurobiol. 2019, 56, 4464–4478.
- Roussakis, A.A.; Gennaro, M.; Gordon, M.F.; Reilmann, R.; Borowsky, B.; Rynkowski, G.; Lao-Kaim, N.P.; Papoutsou, Z.; Savola, J.M.; Hayden, M.R.; et al. A PET-CT study on neuroinflammation in Huntington’s disease patients participating in a randomized trial with laquinimod. Brain Commun. 2023, 5, fcad084.
- Bonelli, R.M.; Heuberger, C.; Reisecker, F. Minocycline for Huntington’s disease: An open label study. Neurology 2003, 60, 883–884.
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801.
- Huntington Study Group, D.I. A futility study of minocycline in Huntington’s disease. Mov. Disord. 2010, 25, 2219–2224.
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER Stress-Induced eIF2-alpha Phosphorylation Underlies Sensitivity of Striatal Neurons to Pathogenic Huntingtin. PLoS ONE 2014, 9, e90803.
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008, 22, 3308–3319.
- Carnemolla, A.; Fossale, E.; Agostoni, E.; Michelazzi, S.; Calligaris, R.; De Maso, L.; Del Sal, G.; MacDonald, M.E.; Persichetti, F. Rrs1 Is Involved in Endoplasmic Reticulum Stress Response in Huntington Disease. J. Biol. Chem. 2009, 284, 18167–18173.
- Cho, K.J.; Lee, B.I.; Cheon, S.Y.; Kim, H.W.; Kim, H.J.; Kim, G.W. Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience 2009, 163, 1128–1134.
- Noh, J.-Y.; Lee, H.; Song, S.; Kim, N.S.; Im, W.; Kim, M.; Seo, H.; Chung, C.-W.; Chang, J.-W.; Ferrante, R.J.; et al. SCAMP5 Links Endoplasmic Reticulum Stress to the Accumulation of Expanded Polyglutamine Protein Aggregates via Endocytosis Inhibition. J. Biol. Chem. 2009, 284, 11318–11325.
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262.
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22.
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595.
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262.
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894.
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576.
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178.
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16.
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiro, C.; Millan, J.M.; Vazquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283.
- Su, T.-P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566.
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 2015, 127, 17–29.
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J Cell Mol Med. 2015, 19, 2540–2548.
- de Yebenes, J.G.; Landwehrmeyer, B.; Squitieri, F.; Reilmann, R.; Rosser, A.; Barker, R.A.; Saft, C.; Magnet, M.K.; Sword, A.; Rembratt, A.; et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011, 10, 1049–1057.
- Huntington Study Group, H.I. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov. Disord. 2013, 28, 1407–1415.
- Lundin, A.; Dietrichs, E.; Haghighi, S.; Goller, M.L.; Heiberg, A.; Loutfi, G.; Widner, H.; Wiktorin, K.; Wiklund, L.; Svenningsson, A.; et al. Efficacy and safety of the dopaminergic stabilizer Pridopidine (ACR16) in patients with Huntington’s disease. Clin. Neuropharmacol. 2010, 33, 260–264.
- Reilmann, R.; McGarry, A.; Grachev, I.D.; Savola, J.-M.; Borowsky, B.; Eyal, E.; Gross, N.; Langbehn, D.; Schubert, R.; Wickenberg, A.T.; et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): A phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019, 18, 165–176.
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751.
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417.
- Dedeoglu, A.; Kubilus, J.K.; Jeitner, T.M.; Matson, S.A.; Bogdanov, M.; Kowall, N.W.; Matson, W.R.; Cooper, A.J.; Ratan, R.R.; Beal, M.F.; et al. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J. Neurosci. 2002, 22, 8942–8950.
- Verny, C.; Bachoud-Levi, A.C.; Durr, A.; Goizet, C.; Azulay, J.P.; Simonin, C.; Tranchant, C.; Calvas, F.; Krystkowiak, P.; Charles, P.; et al. A randomized, double-blind, placebo-controlled trial evaluating cysteamine in Huntington’s disease. Mov. Disord. 2017, 32, 932–936.
- Cherny, R.A.; Ayton, S.; Finkelstein, D.I.; Bush, A.I.; McColl, G.; Massa, S.M. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2012, 1, 211–219.
- Huntington Study Group Reach, H.D.I. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 39–47.
- Wojtecki, L.; Groiss, S.J.; Hartmann, C.J.; Elben, S.; Omlor, S.; Schnitzler, A.; Vesper, J. Deep Brain Stimulation in Huntington’s Disease-Preliminary Evidence on Pathophysiology, Efficacy and Safety. Brain Sci. 2016, 6, 38.
- Wojtecki, L.; Groiss, S.J.; Ferrea, S.; Elben, S.; Hartmann, C.J.; Dunnett, S.B.; Rosser, A.; Saft, C.; Sudmeyer, M.; Ohmann, C.; et al. A Prospective Pilot Trial for Pallidal Deep Brain Stimulation in Huntington’s Disease. Front. Neurol. 2015, 6, 177.
- Fasano, A.; Llinas, M.; Munhoz, R.P.; Hlasny, E.; Kucharczyk, W.; Lozano, A.M. MRI-guided focused ultrasound thalamotomy in non-ET tremor syndromes. Neurology 2017, 89, 771–775.
- Kapadia, A.N.; Elias, G.J.B.; Boutet, A.; Germann, J.; Pancholi, A.; Chu, P.; Zhong, J.; Fasano, A.; Munhoz, R.; Chow, C.; et al. Multimodal MRI for MRgFUS in essential tremor: Post-treatment radiological markers of clinical outcome. J. Neurol. Neurosurg. Psychiatry 2020, 91, 921–927.
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147.
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228.
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147.
- Vescovi, A.L.; Gritti, A.; Galli, R.; Parati, E.A. Isolation and intracerebral grafting of nontransformed multipotential embryonic human CNS stem cells. J. Neurotrauma 1999, 16, 689–693.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872.
- Valor, L.M. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: A genome-wide perspective. Mol. Neurobiol. 2015, 51, 406–423.
- Seredenina, T. and Luthi-Carter, R. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 2012, 45, 83–98.
- Diamanti, D.; Mori, E.; Incarnato, D.; Malusa, F.; Fondelli, C.; Magnoni, L.; Pollio, G. Whole gene expression profile in blood reveals multiple pathways deregulation in R6/2 mouse model. Biomark Res. 2013, 1, 28.
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926.
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum. Mol. Genet. 2005, 14, 1863–1876.
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142.
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977.
- Mitchell, C.T.; Krier, I.; Arjomand, J.; Borowsky, B.; Tabrizi, S.J.; Leavitt, B.R.; Investigators, T.-H.; Luthi-Carter, R. Longitudinal expression changes are weak correlates of disease progression in Huntington’s disease. Brain Commun. 2020, 2, fcaa172.
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A Systematic Review of Transcriptional Dysregulation in Huntington’s Disease Studied by RNA Sequencing. Front. Genet. 2021, 12, 751033.
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020, 8, 19.
- Malaiya, S.; Cortes-Gutierrez, M.; Herb, B.R.; Coffey, S.R.; Legg, S.R.W.; Cantle, J.P.; Colantuoni, C.; Carroll, J.B.; Ament, S.A. Single-Nucleus RNA-Seq Reveals Dysregulation of Striatal Cell Identity Due to Huntington’s Disease Mutations. J. Neurosci. 2021, 41, 5534–5552.
- Lee, H.; Fenster, R.J.; Pineda, S.S.; Gibbs, W.S.; Mohammadi, S.; Davila-Velderrain, J.; Garcia, F.J.; Therrien, M.; Novis, H.S.; Gao, F.; et al. Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 2020, 107, 891–908.e8.
- Rudinskiy, N.; Kaneko, Y.A.; Beesen, A.A.; Gokce, O.; Regulier, E.; Deglon, N.; Luthi-Carter, R. Diminished hippocalcin expression in Huntington’s disease brain does not account for increased striatal neuron vulnerability as assessed in primary neurons. J. Neurochem. 2009, 111, 460–472.
- Seredenina, T.; Gokce, O.; Luthi-Carter, R. Decreased striatal RGS2 expression is neuroprotective in Huntington’s disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS ONE 2011, 6, e22231.
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330.
- Lu, B. BDNF and activity-dependent synaptic modulation. Learn. Mem. 2003, 10, 86–98.
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010, 30, 1739–1749.
- Strand, A.D.; Baquet, Z.C.; Aragaki, A.K.; Holmans, P.; Yang, L.; Cleren, C.; Beal, M.F.; Jones, L.; Kooperberg, C.; Olson, J.M.; et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 2007, 27, 11758–11768.
- Xie, Y.; Hayden, M.R.; Xu, B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 2010, 30, 14708–14718.
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138.
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; van de Bospoort, R.; Veenman-Koepke, J.; et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS ONE 2014, 9, e87923.
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s disease. Neurobiol. Dis. 2009, 36, 259–268.
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768.
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169.
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 2003, 12, 497–508.
- Wyttenbach, A.; Swartz, J.; Kita, H.; Thykjaer, T.; Carmichael, J.; Bradley, J.; Brown, R.; Maxwell, M.; Schapira, A.; Orntoft, T.F.; et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1829–1845.
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428.
- Adachi, M.; Monteggia, L.M. Decoding transcriptional repressor complexes in the adult central nervous system. Neuropharmacology 2014, 80, 45–52.
- Buckley, N.J.; Johnson, R.; Zuccato, C.; Bithell, A.; Cattaneo, E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 2010, 39, 28–39.
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1alpha, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46.
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135.
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101.
- Weydt, P.; Soyal, S.M.; Gellera, C.; Didonato, S.; Weidinger, C.; Oberkofler, H.; Landwehrmeyer, G.B.; Patsch, W. The gene coding for PGC-1alpha modifies age at onset in Huntington’s Disease. Mol. Neurodegener. 2009, 4, 3.
- Taherzadeh-Fard, E.; Saft, C.; Andrich, J.; Wieczorek, S.; Arning, L. PGC-1alpha as modifier of onset age in Huntington disease. Mol. Neurodegener. 2009, 4, 10.
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69.
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362.
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750.
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142.
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55.
- Fink, K.D.; Deng, P.; Gutierrez, J.; Anderson, J.S.; Torrest, A.; Komarla, A.; Kalomoiris, S.; Cary, W.; Anderson, J.D.; Gruenloh, W.; et al. Allele-Specific Reduction of the Mutant Huntingtin Allele Using Transcription Activator-Like Effectors in Human Huntington’s Disease Fibroblasts. Cell Transpl. 2016, 25, 677–686.
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576.
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724.
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847.
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30.
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474.
- Rodriguez-Lebron, E.; Denovan-Wright, E.M.; Nash, K.; Lewin, A.S.; Mandel, R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. 2005, 12, 618–633.
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial microRNAs as siRNA shuttles: Improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2009, 17, 169–175.
- Machida, Y.; Okada, T.; Kurosawa, M.; Oyama, F.; Ozawa, K.; Nukina, N. rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem. Biophys. Res. Commun. 2006, 343, 190–197.
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Mol. Ther. Nucleic Acids. 2016, 5, e297.
- Samaranch, L.; Blits, B.; San Sebastian, W.; Hadaczek, P.; Bringas, J.; Sudhakar, V.; Macayan, M.; Pivirotto, P.J.; Petry, H.; Bankiewicz, K.S. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 2017, 24, 253–261.
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131.
- US Food and Drug Administration. FDA Approves First Drug for Spinal Muscular Atrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy (accessed on 17 August 2023).
- US Food and Drug Administration. FDA Grants Accelerated Approval to First Drug for Duchenne Muscular Dystrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular-dystrophy (accessed on 17 August 2023).
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044.
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J Med. 2019, 380, 2307–2316.
- Roche, Roche-Genentech-HD-Global-Community-Letter-January-2022, 2022. Available online: https://hdsa.org/wp-content/uploads/2022/09/Roche-Global-Patient-Community-letter_EHDN-2022.pdf (accessed on 17 August 2023).
This entry is offline, you can click here to edit this entry!