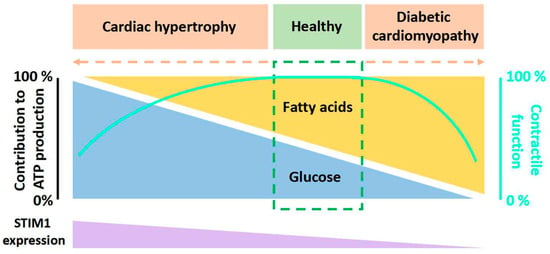
The heart requires a variety of energy substrates to maintain proper contractile function. Glucose and long-chain fatty acids (FA) are the major cardiac metabolic substrates under physiological conditions. Upon stress, a shift of cardiac substrate preference toward either glucose or FA is associated with cardiac diseases. For example, in pressure-overloaded hypertrophic hearts, there is a long-lasting substrate shift toward glucose, while in hearts with diabetic cardiomyopathy, the fuel is switched toward FA. Stromal interaction molecule 1 (STIM1), a well-established calcium (Ca2+) sensor of endoplasmic reticulum (ER) Ca2+ store, is increasingly recognized as a critical player in mediating both cardiac hypertrophy and diabetic cardiomyopathy. However, the cause–effect relationship between STIM1 and glucose/FA metabolism and the possible mechanisms by which STIM1 is involved in these cardiac metabolic diseases are poorly understood.
- STIM1
- cardiac energy metabolism
- cardiac hypertrophy
- diabetic cardiomyopathy
- glucose
- fatty acid
1. Introduction
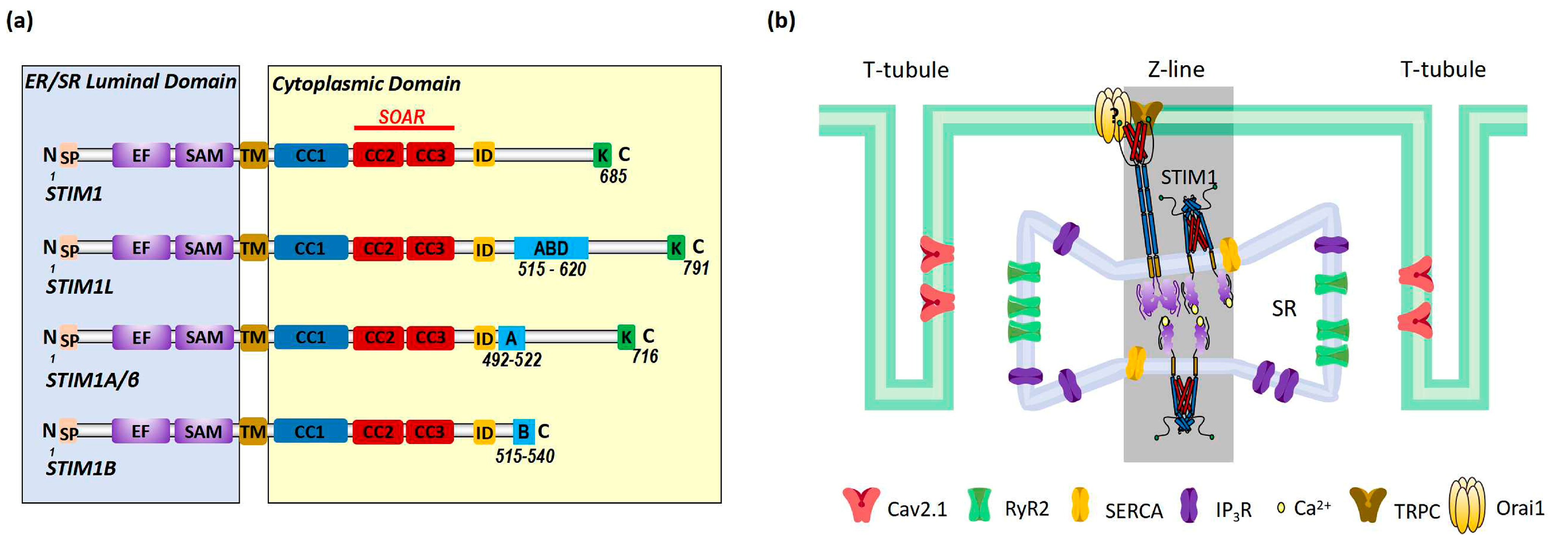
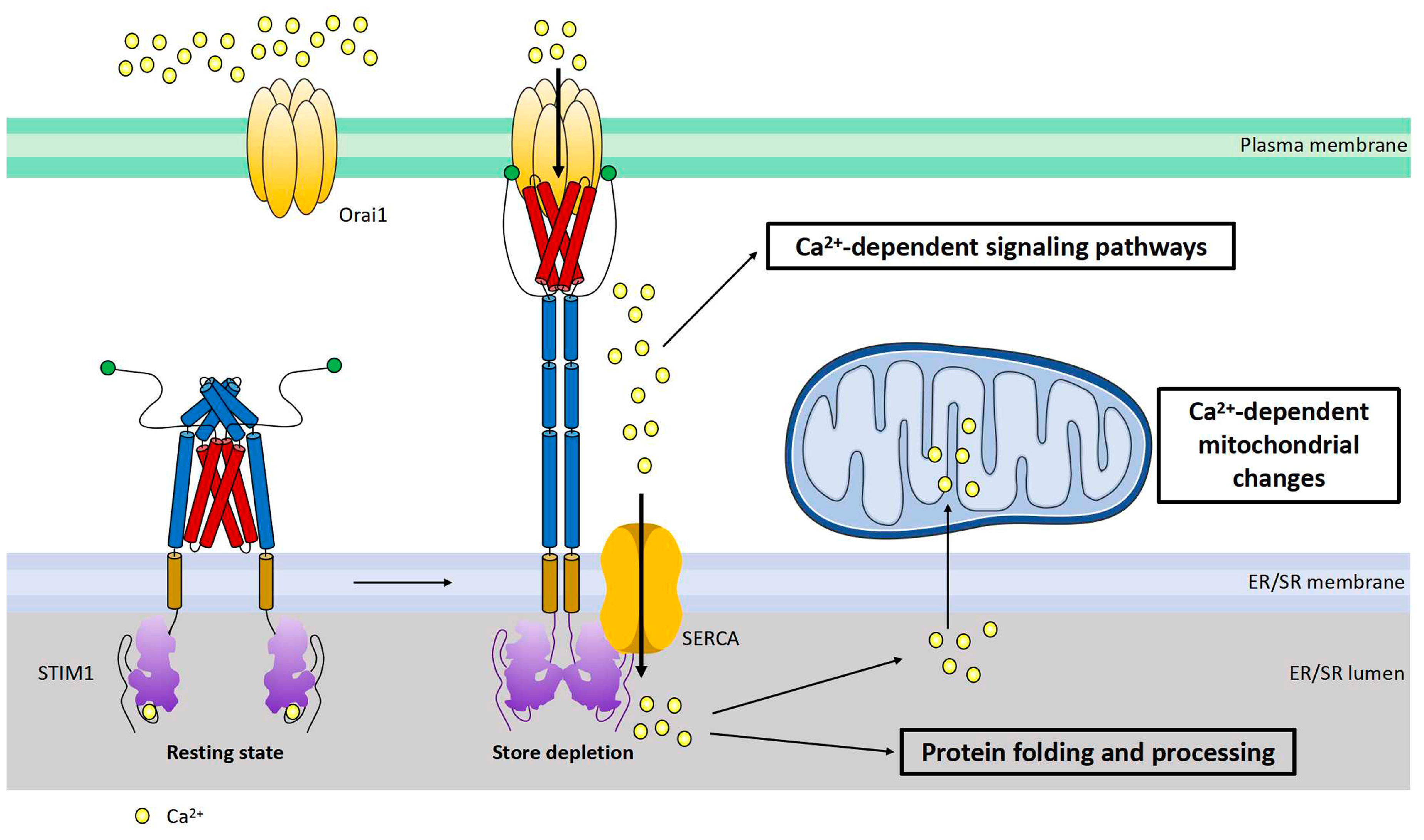
2. STIM1-Dependent Signaling
This entry is adapted from the peer-reviewed paper 10.3390/ijms241713188
References
- Heggermont, W.A.; Papageorgiou, A.P.; Heymans, S.; van Bilsen, M. Metabolic support for the heart: Complementary therapy for heart failure? Eur. J. Heart Fail. 2016, 18, 1420–1429.
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129.
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724.
- Chess, D.J.; Stanley, W.C. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 2008, 79, 269–278.
- Glatz, J.F.C.; Nabben, M.; Young, M.E.; Schulze, P.C.; Taegtmeyer, H.; Luiken, J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim. Biophys. Acta Mol. Basis. Dis. 2020, 1866, 165579.
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368.
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513.
- Liu, P.; Yang, Z.; Wang, Y.; Sun, A. Role of STIM1 in the Regulation of Cardiac Energy Substrate Preference. Int. J. Mol. Sci. 2023, 24, 13188. https://doi.org/10.3390/ijms241713188
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436.
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244.
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826.
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell. Biol. 2012, 13, 549–565.
- van Dorp, S.; Qiu, R.; Choi, U.B.; Wu, M.M.; Yen, M.; Kirmiz, M.; Brunger, A.T.; Lewis, R.S. Conformational dynamics of auto-inhibition in the ER calcium sensor STIM1. eLife 2021, 10, e66194.
- Rathner, P.; Fahrner, M.; Cerofolini, L.; Grabmayr, H.; Horvath, F.; Krobath, H.; Gupta, A.; Ravera, E.; Fragai, M.; Bechmann, M.; et al. Interhelical interactions within the STIM1 CC1 domain modulate CRAC channel activation. Nat. Chem. Biol. 2021, 17, 196–204.
- Shrestha, N.; Hye-Ryong Shim, A.; Maneshi, M.M.; See-Wai Yeung, P.; Yamashita, M.; Prakriya, M. Mapping interactions between the CRAC activation domain and CC1 regulating the activity of the ER Ca2+ sensor STIM1. J. Biol. Chem. 2022, 298, 102157.
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343.
- Knapp, M.L.; Alansary, D.; Poth, V.; Forderer, K.; Sommer, F.; Zimmer, D.; Schwarz, Y.; Kunzel, N.; Kless, A.; Machaca, K.; et al. A longer isoform of Stim1 is a negative SOCE regulator but increases cAMP-modulated NFAT signaling. EMBO Rep. 2022, 23, e53135.
- Xie, J.; Ma, G.; Zhou, L.; He, L.; Zhang, Z.; Tan, P.; Huang, Z.; Fang, S.; Wang, T.; Lee, Y.T.; et al. Identification of a STIM1 Splicing Variant that Promotes Glioblastoma Growth. Adv. Sci. 2022, 9, e2103940.
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwar, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A short isoform of STIM1 confers frequency-dependent synaptic enhancement. Cell Rep. 2021, 34, 108844.
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346.
- Sauc, S.; Bulla, M.; Nunes, P.; Orci, L.; Marchetti, A.; Antigny, F.; Bernheim, L.; Cosson, P.; Frieden, M.; Demaurex, N. STIM1L traps and gates Orai1 channels without remodeling the cortical ER. J. Cell Sci. 2015, 128, 1568–1579.
- Luo, X.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Tandan, S.; Rakalin, A.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2012, 52, 136–147.
- Voelkers, M.; Salz, M.; Herzog, N.; Frank, D.; Dolatabadi, N.; Frey, N.; Gude, N.; Friedrich, O.; Koch, W.J.; Katus, H.A.; et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J. Mol. Cell Cardiol. 2010, 48, 1329–1334.
- Hulot, J.S.; Fauconnier, J.; Ramanujam, D.; Chaanine, A.; Aubart, F.; Sassi, Y.; Merkle, S.; Cazorla, O.; Ouille, A.; Dupuis, M.; et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation 2011, 124, 796–805.
- Correll, R.N.; Goonasekera, S.A.; van Berlo, J.H.; Burr, A.R.; Accornero, F.; Zhang, H.; Makarewich, C.A.; York, A.J.; Sargent, M.A.; Chen, X.; et al. STIM1 elevation in the heart results in aberrant Ca2+ handling and cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 87, 38–47.
- Troupes, C.D.; Wallner, M.; Borghetti, G.; Zhang, C.; Mohsin, S.; von Lewinski, D.; Berretta, R.M.; Kubo, H.; Chen, X.; Soboloff, J.; et al. Role of STIM1 (Stromal Interaction Molecule 1) in Hypertrophy-Related Contractile Dysfunction. Circ. Res. 2017, 121, 125–136.
- Rosenberg, P.; Zhang, H.; Bryson, V.G.; Wang, C. SOCE in the cardiomyocyte: The secret is in the chambers. Pflug. Arch. 2021, 473, 417–434.
- Zhang, H.; Sun, A.Y.; Kim, J.J.; Graham, V.; Finch, E.A.; Nepliouev, I.; Zhao, G.; Li, T.; Lederer, W.J.; Stiber, J.A.; et al. STIM1-Ca2+ signaling modulates automaticity of the mouse sinoatrial node. Proc. Natl. Acad. Sci. USA 2015, 112, E5618–E5627.
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca(2+) handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224.
- Zhao, G.; Li, T.; Brochet, D.X.; Rosenberg, P.B.; Lederer, W.J. STIM1 enhances SR Ca2+ content through binding phospholamban in rat ventricular myocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E4792–E4801.
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Adv. Exp. Med. Biol. 2017, 993, 505–521.
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126.
- Gokce, Y.; Erkan, O.; Savas, K.; Rahman, T.; Yaras, N. Pharmacological blockade of angiotensin II receptor restores diabetes-associated reduction of store operated Ca2+ entry in adult cardiomyocytes. Biochem. Biophys. Res. Commun. 2022, 610, 56–60.
- Shen, W.W.; Frieden, M.; Demaurex, N. Remodelling of the endoplasmic reticulum during store-operated calcium entry. Biol. Cell 2011, 103, 365–380.
- McIvor, E.; Coombes, S.; Thul, R. Three-dimensional spatio-temporal modelling of store operated Ca2+ entry: Insights into ER refilling and the spatial signature of Ca2+ signals. Cell Calcium 2018, 73, 11–24.
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 11456–11464.
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflug. Arch. 2018, 470, 1555–1567.
- Estrada, I.A.; Donthamsetty, R.; Debski, P.; Zhou, M.H.; Zhang, S.L.; Yuan, J.X.; Han, W.; Makino, A. STIM1 restores coronary endothelial function in type 1 diabetic mice. Circ. Res. 2012, 111, 1166–1175.
- Collins, H.E.; He, L.; Zou, L.; Qu, J.; Zhou, L.; Litovsky, S.H.; Yang, Q.; Young, M.E.; Marchase, R.B.; Chatham, J.C. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1231–H1239.
- Takeuchi, A.; Matsuoka, S. Spatial and Functional Crosstalk between the Mitochondrial Na+-Ca2+ Exchanger NCLX and the Sarcoplasmic Reticulum Ca2+ Pump SERCA in Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 7948.
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529.
- Kraus, W.E.; Muoio, D.M.; Stevens, R.; Craig, D.; Bain, J.R.; Grass, E.; Haynes, C.; Kwee, L.; Qin, X.; Slentz, D.H.; et al. Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis. PLoS Genet. 2015, 11, e1005553.
- Dhande, I.S.; Zhu, Y.; Kneedler, S.C.; Joshi, A.S.; Hicks, M.J.; Wenderfer, S.E.; Braun, M.C.; Doris, P.A. Stim1 Polymorphism Disrupts Immune Signaling and Creates Renal Injury in Hypertension. J. Am. Heart Assoc. 2020, 9, e014142.
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338.
- Zhang, X.; Liu, S.; Zhang, G.; Zhong, M.; Liu, T.; Wei, M.; Wu, D.; Huang, X.; Cheng, Y.; Wu, Q.; et al. Bariatric Surgery Ameliorates Diabetic Cardiac Dysfunction by Inhibiting ER Stress in a Diabetic Rat Model. Obes. Surg. 2017, 27, 1324–1334.
- Yuan, M.; Gong, M.; Zhang, Z.; Meng, L.; Tse, G.; Zhao, Y.; Bao, Q.; Zhang, Y.; Yuan, M.; Liu, X.; et al. Hyperglycemia Induces Endoplasmic Reticulum Stress in Atrial Cardiomyocytes, and Mitofusin-2 Downregulation Prevents Mitochondrial Dysfunction and Subsequent Cell Death. Oxid. Med. Cell. Longev. 2020, 2020, 6569728.
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169.
- Tian, J.H.; Wu, Q.; He, Y.X.; Shen, Q.Y.; Rekep, M.; Zhang, G.P.; Luo, J.D.; Xue, Q.; Liu, Y.H. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 2021, 42, 393–403.
- Balaban, R.S. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J. Mol. Cell. Cardiol. 2002, 34, 1259–1271.
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49.
- Houten, S.M.; Chegary, M.; Te Brinke, H.; Wijnen, W.J.; Glatz, J.F.; Luiken, J.J.; Wijburg, F.A.; Wanders, R.J. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell. Mol. Life Sci. 2009, 66, 1283–1294.
- Wong, A.K.; Howie, J.; Petrie, J.R.; Lang, C.C. AMP-activated protein kinase pathway: A potential therapeutic target in cardiometabolic disease. Clin. Sci. 2009, 116, 607–620.
- Graves, B.M.; Simerly, T.; Li, C.; Williams, D.L.; Wondergem, R. Phosphoinositide-3-kinase/akt—dependent signaling is required for maintenance of i/Ca, and Ca2+ transients in HL-1 cardiomyocytes. J. Biomed. Sci. 2012, 19, 59.
- Angin, Y.; Schwenk, R.W.; Nergiz-Unal, R.; Hoebers, N.; Heemskerk, J.W.; Kuijpers, M.J.; Coumans, W.A.; van Zandvoort, M.A.; Bonen, A.; Neumann, D.; et al. Calcium signaling recruits substrate transporters GLUT4 and CD36 to the sarcolemma without increasing cardiac substrate uptake. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E225–E236.
- Schonekess, B.O.; Brindley, P.G.; Lopaschuk, G.D. Calcium regulation of glycolysis, glucose oxidation, and fatty acid oxidation in the aerobic and ischemic heart. Can. J. Physiol. Pharmacol. 1995, 73, 1632–1640.
- Kusuhara, K.; Madsen, K.; Jensen, L.; Hellsten, Y.; Pilegaard, H. Calcium signalling in the regulation of PGC-1alpha, PDK4 and HKII mRNA expression. Biol. Chem. 2007, 388, 481–488.
- Xie, Y.; Gu, Z.J.; Wu, M.X.; Huang, T.C.; Ou, J.S.; Ni, H.S.; Lin, M.H.; Yuan, W.L.; Wang, J.F.; Chen, Y.X. Disruption of calcium homeostasis by cardiac-specific over-expression of PPAR-gamma in mice: A role in ventricular arrhythmia. Life Sci. 2016, 167, 12–21.
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211.
- Grigoriev, I.; Gouveia, S.M.; van der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W., Jr.; Hoogenraad, C.C.; et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 2008, 18, 177–182.
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109.
- Krapivinsky, G.; Krapivinsky, L.; Stotz, S.C.; Manasian, Y.; Clapham, D.E. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 19234–19239.
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438.
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347.
- Gammons, J.; Halpage, J.; Mancarella, S. Mapping the Proximity Interaction Network of STIM1 Reveals New Mechanisms of Cytoskeletal Regulation. Cells 2021, 10, 2701.
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162.
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75.
- Xu, K.Y.; Becker, L.C. Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum vesticles. J. Histochem. Cytochem. 1998, 46, 419–427.
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim Biophys Acta 2010, 1797, 607–618.
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1068–1078.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345.
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356.
- Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946.
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72.
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405.
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 2012, 287, 42042–42052.
- Li, B.; Xiao, L.; Wang, Z.Y.; Zheng, P.S. Knockdown of STIM1 inhibits 6-hydroxydopamine-induced oxidative stress through attenuating calcium-dependent ER stress and mitochondrial dysfunction in undifferentiated PC12 cells. Free Radic. Res. 2014, 48, 758–768.
- Nan, J.; Li, J.; Lin, Y.; Saif Ur Rahman, M.; Li, Z.; Zhu, L. The interplay between mitochondria and store-operated Ca2+ entry: Emerging insights into cardiac diseases. J. Cell. Mol. Med. 2021, 25, 9496–9512.
- Yang, D.; Liu, H.Q.; Liu, F.Y.; Guo, Z.; An, P.; Wang, M.Y.; Yang, Z.; Fan, D.; Tang, Q.Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2021, 8, 822969.
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53.
- Galloway, C.A.; Yoon, Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1545–1562.
- Cai, C.; Wu, F.; He, J.; Zhang, Y.; Shi, N.; Peng, X.; Ou, Q.; Li, Z.; Jiang, X.; Zhong, J.; et al. Mitochondrial quality control in diabetic cardiomyopathy: From molecular mechanisms to therapeutic strategies. Int. J. Biol. Sci. 2022, 18, 5276–5290.
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195.
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808.
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328.
- Givvimani, S.; Munjal, C.; Tyagi, N.; Sen, U.; Metreveli, N.; Tyagi, S.C. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE 2012, 7, e32388.
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; Del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127 Pt 12, 2659–2671.
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes 2014, 63, 75–88.
- Gao, Q.; Wang, X.M.; Ye, H.W.; Yu, Y.; Kang, P.F.; Wang, H.J.; Guan, S.D.; Li, Z.H. Changes in the expression of cardiac mitofusin-2 in different stages of diabetes in rats. Mol. Med. Rep. 2012, 6, 811–814.
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R., 3rd; Hoshijima, M.; Dillmann, W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 2012, 287, 30024–30034.
- Collins, H.E.; Pat, B.M.; Zou, L.; Litovsky, S.H.; Wende, A.R.; Young, M.E.; Chatham, J.C. Novel role of the ER/SR Ca2+ sensor STIM1 in the regulation of cardiac metabolism. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1014–H1026.
- Singaravelu, K.; Nelson, C.; Bakowski, D.; de Brito, O.M.; Ng, S.W.; Di Capite, J.; Powell, T.; Scorrano, L.; Parekh, A.B. Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J. Biol. Chem. 2011, 286, 12189–12201.
- Nunes, P.; Demaurex, N. Redox regulation of store-operated Ca2+ entry. Antioxid. Redox Signal. 2014, 21, 915–932.
- Vaeth, M.; Maus, M.; Klein-Hessling, S.; Freinkman, E.; Yang, J.; Eckstein, M.; Cameron, S.; Turvey, S.E.; Serfling, E.; Berberich-Siebelt, F.; et al. Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 2017, 47, 664–679 e6.