Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Growing evidence supports the view that statins, ezetimibe, PCSK9 inhibitors, inclisiran, and icosapent ethyl also act as antithrombotics. The main effect of antidyslipidemic agents is mainly related to the reduction in low-density lipoprotein (LDL) levels (which are causally related to atherosclerosis) and triglycerides. Some studies suggested a potential role of these drugs also on platelet function. In particular, by interacting with specific platelet receptors, they reduce adhesion, aggregation, degranulation, in-flammation, vasoconstriction, and oxidative stress.
- platelet
- residual platelet reactivity
- anticholesterolemic drugs
- statin
- ezetimibe
- PCSK9i
- inclisiran
- icosapent ethyl
1. Introduction
Atherosclerosis is the primary cause of cardiovascular deaths, regardless of gender [1]. It involves the gradual development of fatty streaks in arterial walls, which transform into atheroma and eventually into vulnerable plaques. These atherosclerotic plaques consist of a fibrous cap and a lipid core, primarily composed of oxidized low-density lipoprotein (Ox-LDL). The plaque’s stability relies mainly on the fibrous cap’s thickness (FCT), as the exposure of the core material to the blood can trigger platelet activation. A vulnerable plaque is defined as having an FCT of ≤65 μm. Plaque rupture and subsequent formation of an intraluminal thrombus are the most common causes of acute coronary syndromes [2].
It has been established that nonpharmacological and pharmacological interventions aimed at lowering cholesterol levels impact cardiovascular morbidity and mortality substantially. The history of hypercholesterolemic therapy starts in 1950 with the description of the cholesterol synthesis pathway. The evolution of hypolipidemic therapy has made it possible to achieve ambitious therapeutic targets in a short time.
The direct effect of the drugs currently available is related to lowering LDL-C. Several studies have shown an average cardiovascular risk decrease of 22–23% per 1.0 mmol/L LDL-C reduction [3]. Statins inhibit HMG-CoA reductase, causing the reduction in endogenous cholesterol synthesis in the liver. Ezetimibe inhibits the absorption of dietary and biliary cholesterol by inhibiting the Niemann–Pick C1-like 1 (NPC1L1) protein expressed in intestinal cells and hepatocytes. PCSK9 is a hepatic protease that binds to the LDLR receptor on the surface of the liver and causes its degradation. PCSK9 inhibitors are monoclonal antibodies that reduce the expression of this protease, leading to increased LDL-C uptake. Inclisiran is a small interfering RNA (siRNA) that penetrates hepatocytes and blocks the translation of PCSK9 mRNA. Bempedoic acid acts similarly to statins by blocking cholesterol synthesis through the inhibition of adenosine triphosphate citrate lyase (ACLY). The rationale for the use of hypolipidemic drugs is mainly related to the stabilizing effect of atheromatous plaque, the decrease in the amount of the lipid core, and the increase in the thickness of the fibrous cap (Figure 1) [4].
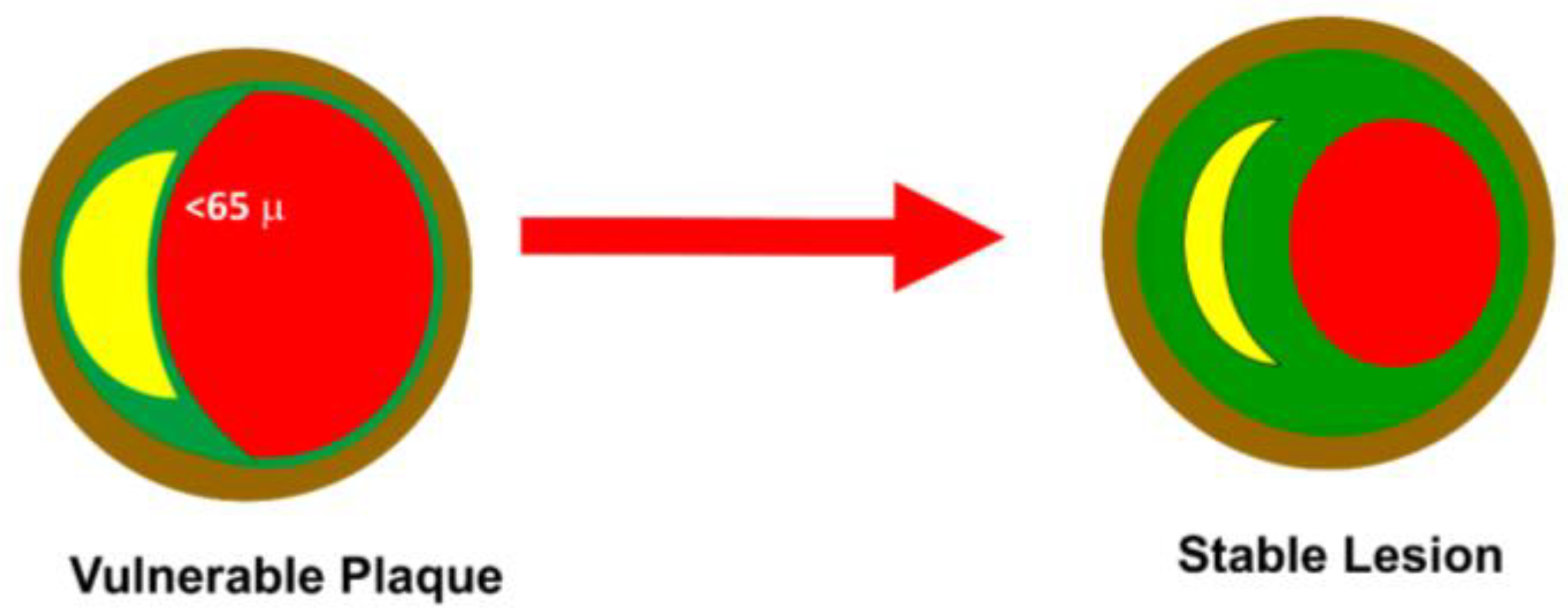
Figure 1. Plaque stability. A vulnerable plaque is defined as having an FCT of ≤65 μm with a pronounced lipid core. The anticholesterolemic drugs work through a reduction in lipid core and an increase in FCT. The severity of the lesion usually does not impact the transition from stable to unstable plaque.
As a result, the latest international lipid guidelines now strongly advocate for lipid-lowering therapy as a crucial component of primary and secondary prevention in patients with atherosclerotic cardiovascular disease (ASCVD). In addition to diet and maximum tolerated statin or ezetimibe therapy, several additional lipid-lowering medications are available, including two fully human monoclonal antibodies (mAbs) anti-proprotein convertase subtilisin/kexin type 9 (PCSK9), alirocumab and evolocumab, a small interfering RNA (siRNA) that prevents the hepatic synthesis of PCSK9, inclisiran, and a further novel nonstatin drug bempedoic acid, are recently approved as agents available for use in adult hypercholesterolemia who do not achieve target LDL-C [5].
2. Interaction between Atherosclerotic Plaque and Platelet Activity
In vivo and in vitro studies have shown that platelets adhere to the intact endothelium without proaggregating factors [6]. Endothelium-expressed P-selectin (CD62P) contacts the platelet receptor PSGL-1 (P-selectin glycoprotein ligand-1) and mediates reversible adhesion and rolling. Stable adhesion is mediated by integrins, a family of CAMs (cell adhesion molecules). Stable binding to the endothelium activates the formation of an inflammatory environment that predisposes the development of atherosclerotic lesions. Platelets firmly attached to the endothelium increase the release of CD40L on the platelet surface, which then releases soluble CD40L. Activated platelets also express P-selectin. CD40L binding on the surface of platelets and endothelium increases adhesion molecules such as E-selectin, VCAM-1, ICAM-1, and proinflammatory cytokines such as interleukin-8 (IL-8), interleukin (IL)-6, RANTES (regulated on activation normal T cells expressed and secreted), monocyte endothelial chemotactic protein-1 (MCP-1), and metalloproteinase (MMP). MMPs are involved in the remodeling and degradation of the extracellular matrix. In addition, platelets release IL-1β, which increases MCP-1 secretion. MCP-1 protein promotes the recruitment of monocytes, which adhere to the endothelium through the platelet chemotactic factor RANTES and binding P-selectins and, subsequently the integrins VCAM (vascular cell adhesion molecule) [7]. Activated platelets on the endothelial surface release alpha-granules and secrete PF4 (platelet factor 4), among other proteins. PF4 is involved in the differentiation of monocytes into macrophages. Furthermore, it was observed that PF4 and Ox-LDL were colocalized at atherosclerotic lesions. PF4 prevents LDL from binding to the macrophage receptor LDL-R. In this way, LDL is not degraded but remains trapped in the vascular space and is oxidized [8][9]. Ox-LDL then binds class A scavenger receptors (SR-A, typical of activated macrophages and smooth muscle cells), class B scavenger receptors (CD36, expressed on macrophages and cells in the liver, brain, and heart), and a third class typically expressed by lysosomes (CD68, expressed on cells associated with the immune system and bone marrow such as monocytes, macrophages, dendritic cells, and osteoclasts). These binding favors endocytosis and the formation of foam cells that promote plaque formation and growth [10]. In turn, Ox-LDL increases the local production of chemokines that attract monocytes and the production of oxidized LDL lectin-like receptor-1 (LOX-1). LOX-1 is expressed on endothelial cells and allows Ox-LDL accumulation (Figure 2) [11].
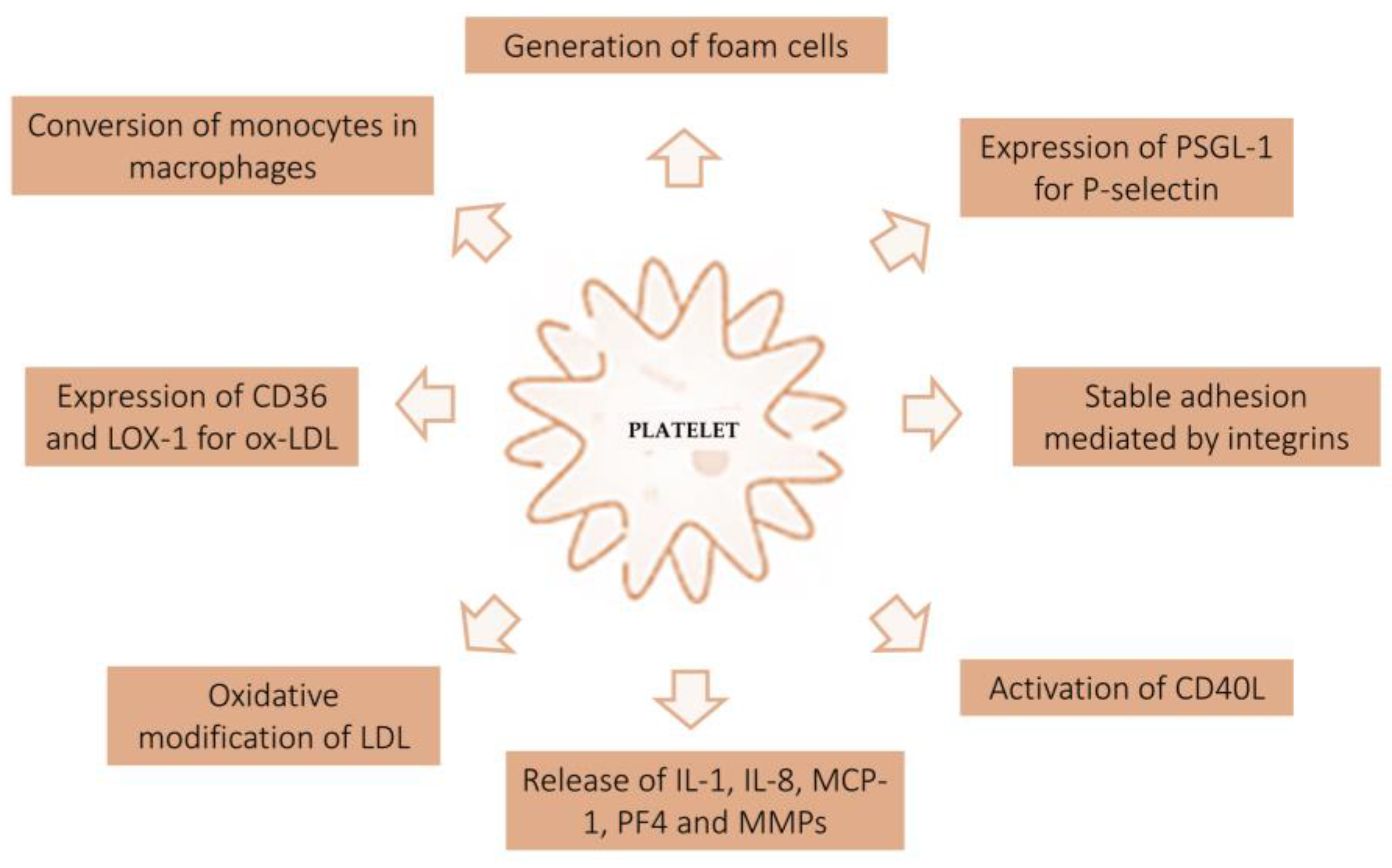
Figure 2. Interaction between atherosclerotic plaque and platelet activity. The figure shows the mechanism of platelet activation at the endothelial level. Based on in vitro and in vivo studies, platelets play a key role in the genesis of atherosclerotic plaque. PSGL-1, P-selectin glycoprotein ligand-1; PF4, platelet factor 4; MCP-1, endothelial monocyte chemotactic protein-1; MMPs, metalloproteinases; LOX-1, lectin-like receptor-1.
Several factors play a role in modulating the platelet response. It has been shown that inflammatory diseases can lead to alterations in platelet function [12]. Indeed, platelets play a major role in the pathogenesis of acute coronary syndromes. The most frequent cause of acute coronary syndrome (ACS) is plaque rupture (60%). The second mechanism of ACS is the erosion of fibrous plaques. In both cases, exposure of the lipid core to the blood is the initial mechanism of platelet aggregation and intracoronary thrombus formation (Figure 3).
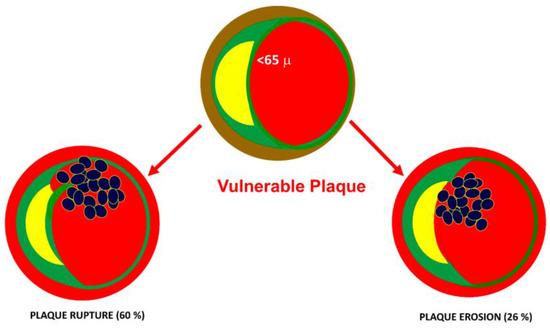
Figure 3. Underlying causes of acute coronary syndromes. The most frequent cause of acute coronary syndrome (ACS) is plaque rupture (60%). The second mechanism of ACS is the erosion of fibrous caps. In both cases, the exposure of the lipid core to the blood is the initial mechanism of platelet aggregation and intracoronary thrombus formation.
Exposure to collagen and von Willebrand factor (vWF) results in platelet activation. Platelets adhere to the endothelium-expressed P-selectin via the platelet glycoprotein (GP) GPIbα and PSGL-1. Subsequently, stable adhesion is mediated by integrins to collagen. The attached platelets release ADP and thromboxane A2. The injured endothelium exposes tissue factors that trigger the coagulation cascade and thrombin activation. Platelet aggregation and thrombus formation are the results of the activation of three signal pathways on the surface of platelets: thrombin protease-activated receptor-1, TxA2-thromboxane receptor, and ADP-P2Y12 receptor pathways. In this way, numerous platelets are drawn to the site of injury, promoting thrombus formation [13][14][15].
3. The Effect of Statins on Platelet Function
The protective effect of statins on cardiovascular events is well-known. Statins, or HMG-CoA reductase inhibitors, have as their main mechanism of action the reduction in endogenous cholesterol synthesis in the liver. The mechanism underlying the beneficial effect is mainly related to reducing the lipid core and increasing the fibrous cap (Figure 1). However, several studies have shown that the protective effect of statins does not depend entirely on LDL-C reduction but is associated with a pleiotropic action. At high doses, statins have been shown in experimental studies performed to reduce neointimal proliferation after vascular injury and left ventricular hypertrophy (LVH) in a pressure overload model [16][17][18]. Restenosis is one of the main mechanisms associated with percutaneous coronary intervention (PCI) failure of bare metal stents (BMS). Coronary stent placement triggers an inflammatory reaction and the differentiation of vascular smooth muscle cells (VSMCs), which migrate and form neointima. Intimal hyperplasia is associated with activation of the ras pathway. Ras protein is responsible for RAF and MAPKK activation (mitogen-activated protein kinase).
In addition, statins are involved in modulating platelet activation. A 2003 study showed that discontinuation of statin therapy was associated with an increase in cardiovascular events with an increase in Ox-LDL, P-selectin, and platelet aggregation observed 14 days after discontinuation [19].
Another mechanism underlying the antiplatelet effect of statins is iteration with different signal transduction pathways. Statins, in particular simvastatin, atorvastatin, and rosuvastatin, play an important role in the inhibition to activation of several pathways (Table 1).
Table 1. Statin interaction in platelet function. The table summarizes the main interactions between platelet activity and the mechanism of action of statins. Inhibitory pathways are indicated in the red box. Activating mechanisms are indicated in the green box.
| Inhibition Pathway | |
| P-selectin | Reduction in platelet stable adhesion |
| PAR-1 | Reduction in platelet aggregation, release of proinflammatory particles, and production of adhesion molecules |
| TF | Reduction in extrinsic coagulation pathway and cMP release |
| CD40L | Reduction in serum inhibits platelet aggregation |
| NOX2/NADPH | Antioxidant effect |
| Activation Pathway | |
| PPARα/PPARƴ | Inhibition of platelet degranulation and aggregation by the suppression of PKCα pathway |
| Increase in cAMP and decrease in Ca2+ level | |
| eNOS | NO has vasoprotective, vasodilating effects, and reduced platelet aggregation |
| PLA2 | Reduction in TxA2 |
PAR-1, protease-activated receptor-1; TF, tissue factor; cMP, circulating microparticles; NOX2, NADPH, PPARα/PPARƴ, peroxisome proliferator-activated receptor; PKCα, protein kinase Cα; cAMP, cyclic adenosine 3′,5′-monophosphate; eNOS, endothelial nitric oxide synthase; PLA2, phospholipase A2; TxA2, thromboxane A2.
4. The Effect of Ezetimibe on Platelet Function
The mechanism of action of statins results in the inhibition of HMG-CoA reductase. This inhibition results in increased expression of hepatic LDL-C receptors and reduction in LDL-C. Intestinal feedback is therefore activated, resulting in increased intestinal absorption of dietary cholesterol. Ezetimibe inhibits the uptake of dietary and biliary cholesterol. The drug’s target is the Niemann–Pick C1-like 1 (NPC1L1) protein expressed in intestinal cells and hepatocytes. At the intestinal level, it reduces the absorption of exogenous cholesterol. At the hepatic level, the reduced absorption of cholesterol increases the expression of LDL receptors with increased hepatic uptake of LDL-C. The effect is a reduction in circulating cholesterol by approximately 10–15%. The addition of ezetimibe in therapy has the advantage of reaching lower LDL-C targets faster and also allows lower statin dosages to be used.
The results of the IMPROVE-IT study demonstrated the efficacy of statin/ezetimibe combination therapy compared to monotherapy. A reduction in LDL-C levels and a further 9% reduction in cardiovascular events (cardiovascular death, acute myocardial infarction, and stroke) were observed one year after randomization [20]. In vitro, studies have demonstrated a direct effect of ezetimibe on endothelium and platelets [21][22]. Ezetimibe directly attenuates platelet activation and has significant endothelial cell-mediated effects on selected markers of atherosclerosis. In particular, ezetimibe has been shown to reduce the expression of urokinase-type plasminogen activator receptor (uPAR or CD87) on endothelial cells [23]. The uPAR plays a key role in the mechanism of platelet adhesion and aggregation: uPAR/uPA binding (expressed by platelets) results in fibrin production and thrombus formation.
5. The Effect of PCSK9i on Platelet Function
PCSK9 is a protease involved in cholesterol metabolism in the liver [24]. Hepatocytes secrete PCSK9, which binds the LDLR receptor on the liver surface and causes its degradation. This leads to an increase in LDL-C cholesterol. PCSK9 inhibitors are monoclonal antibodies that reduce the expression of this protease, leading to a reduction in LDL-C cholesterol by about 60% and a reduction in major cardiovascular adverse events [25][26][27][28]. However, PCSK9 is produced and secreted by other cells: intestinal, pancreatic, adipocytes, kidney, and brain cells. Several recent studies have evaluated the involvement of PCSK9 in cardiovascular disease, independent of increased LDL-C [29]. High serum levels of PCSK9 were indicative of atherosclerotic disease. The clinical implication of this observation was an increased risk of ischemic events, even in subjects with low LDL-C levels. Interesting data demonstrate a correlation between increased serum PCSK9 and platelet reactivity. The PCSK9-REACT study described the association between PCSK9 and platelet activity in ACS patients treated with DAPT. High PCSK9 levels were associated with increased platelet reactivity (p = 0.004) and reduction in the effects of antiplatelet drugs.
The Odyssey and Fourier studies demonstrated the direct effect of PCSK9i (alirocumab and evolocumab, respectively): the reduction in circulating LDL levels by 60%, independent of other hypolipidemic therapies taken by the patient. Moreover, both studies demonstrated a reduction in the primary endpoint in patients with high cardiovascular risk. Alirocumab and evolocumab demonstrated a reduction in cardiovascular events of 15% and 20%, respectively [30][31].
The production of PCSK9 and Ox-LDL is increased in subjects with hypercholesterolemia. High circulating levels of PCSK9 activate platelet scavenger receptors CD36, which induce platelet activation via src and JNK kinases involved in the mechanism of thromboxane A2 production. PCSK9 increases ROS production via NOX2 activation on platelets. This increases Ox-LDL formation, which amplifies platelet activation via binding to the CD36 and LOX1 receptors. This triggers platelet aggregation and thrombogenesis through the expression of p-selectin, CD40L, and granule release. PCSK9is have been shown to reduce the activation of NOX2, CD36, and LOX1. In particular, they demonstrated a reduction in oxidative stress by reducing the NOX2 pathway [32].
6. The Effect of Inclisiran on Platelet Function
Inclisiran is a small interfering RNA (siRNA) directed against PCSK9. Unlike monoclonal antibodies, inclisiran penetrates hepatocytes and blocks the translation of PCSK9 mRNA. Consequently, more LDL receptors will be expressed on the surface of hepatocytes, removing LDL from the bloodstream.
The ORION 9, 10, and 11 studies demonstrated a reduction in LDL-C of about 50% with only two administrations per year [33]. In addition, the ongoing ORION 4 and 5 studies are evaluating the impact of inclisiran on cardiovascular outcomes. The drug is administered subcutaneously, and the only reported adverse events are redness and itching at the injection site. The ORION-1 safety analysis showed that the drug did not cause immunogenicity towards platelets, immune system cells (lymphocytes and monocytes), and immune markers (IL-6 and TNF-α) [34].
PCSK9 is also involved in atherogenic mechanisms, independently of its action on LDL. This protein is involved in proinflammatory and prothrombotic effects.
7. The Effect of Triglycerides on Platelet Function
High triglyceride (TG) levels are a cardiovascular risk factor. Defects in genes controlling TG metabolism, such as APOC3, ANGPTL3, and ANGPTL4, have been linked to reduced TG and reduced CVD risk. In fact, in the Framingham Offspring Study, borderline elevated TG levels > 150 mg/dL were independently associated with a 10–20% increase in CVD [35]. The results of the retrospective TG-Real study assessed an increase in cardiovascular events and death in patients with elevated TG levels [36].
Polyunsaturated fatty acids (PUFA) form the basis of the therapeutic strategy for hypertriglyceridemia. In particular, PUFA-n3 (OMEGA 3) has demonstrated multiple effects, with anti-inflammatory and antithrombotic properties. An inverse relationship was observed between carotid intima-media thickness and PUFA-n3 use. The main components of PUFA-n3 are EPA (eicosapentaenoic acid) and DHA (docosahexaenoic acid). PUFA-n3 formulations are indicated, in combination with diet, to reduce serum triglyceride levels. Icosapent ethyl (IPE) is a drug recently approved by the FDA for cardiovascular prevention in subjects with serum triglycerides > 150 mg/dL. Its composition is 96% purified and stabilized ester of EPA. EPA and IPE have been shown to reduce CV events [37][38][39], in contrast to DHA which can cause increased LDL levels [40].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241411739
References
- Calabrò, P.; Niccoli, G.; Gragnano, F.; Grove, E.L.; Vergallo, R.; Mikhailidis, D.P.; Patti, G.; Spaccarotella, C.; Katsiki, N.; Masiero, G.; et al. Are we ready for a gender-specific approach in interventional cardiology? Int. J. Cardiol. 2018, 286, 226–233.
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411.
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Collins, R. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomized trials. Lancet 2010, 376, 1670–1681.
- Calabrò, P.; Spaccarotella, C.; Cesaro, A.; Andò, G.; Piccolo, R.; De Rosa, S.; Zimarino, M.; Mancone, M.; Gragnano, F.; Moscarella, E.; et al. Lipid-lowering therapy in patients with coronary artery disease undergoing percutaneous coronary interventions in Italy: An expert opinion paper of Interventional Cardiology Working Group of Italian Society of Cardiology. J. Cardiovasc. Med. 2023, 24, e86–e94.
- Calabrò, P.; De Ferrari, G.M.; Romeo, F.; Indolfi, C.; Filardi, P.P. Perrone Filardi, Dalla rivoluzione delle statine alla terapia di silenziamento genico: 50 anni di evoluzione nella terapia dell’ipercolesterolemia. G. Ital. Cardiol. 2022, 23, 481–490.
- Massberg, S.; Brand, K.; Gruner, S.; Page, S.; Muller, E.; Muller, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 2002, 196, 887–896.
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384.
- Nassar, T.; Sachais, B.S.; Akkawi, S.; Kowalska, M.A.; Bdeir, K.; Leitersdorf, E.; Hiss, E.; Ziporen, L.; Aviram, M.; Cines, D.; et al. platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. J. Biol. Chem. 2003, 278, 6187–6193.
- Sachais, B.S.; Kuo, A.; Nassar, T.; Morgan, J.; Kariko, K.; Williams, K.J.; Feldman, M.; Aviram, M.; Shah, N.; Jarett, L.; et al. Platelet factor 4 binds to low-density lipoprotein receptors and disrupts the endocytic itinerary, resulting in retention of low-density lipoprotein on the cell surface. Blood 2002, 99, 3613–3622.
- Steinberg, D. Low Density Lipoprotein Oxidation and Its Pathobiological Significance. J. Biol. Chem. 1997, 272, 20963–20966.
- Dasagrandhi, D.; Muthuswamy, A.; Swaminathan, J.K. Atherosclerosis: Nexus of vascular dynamics and cellular cross talks. Mol. Cell. Biochem. 2021, 477, 571–584.
- Polimeni, A.; Leo, I.; Spaccarotella, C.; Mongiardo, A.; Sorrentino, S.; Sabatino, J.; De Rosa, S.; Indolfi, C. Differences in coagulopathy indices in patients with severe versus non-severe COVID-19: A meta-analysis of 35 studies and 6427 patients. Sci. Rep. 2021, 11, 10464.
- Gurbel, P.A.; Jeong, Y.-H.; Navarese, E.P.; Tantry, U.S. Platelet-Mediated Thrombosis. Circ. Res. 2016, 118, 1380–1391.
- Wagner, D.D.; Burger, P.C. Platelets in Inflammation and Thrombosis. Arter. Thromb. Vasc. Biol. 2003, 23, 2131–2137.
- Massberg, S.; Schulz, C.; Gawaz, M. Role of Platelets in the Pathophysiology of Acute Coronary Syndrome. Semin. Vasc. Med. 2003, 3, 147–162.
- Déglise, S.; Bechelli, C.; Allagnat, F. Vascular smooth muscle cells in intimal hyperplasia, an update. Front. Physiol. 2023, 13, 1081881.
- Indolfi, C.; Cioppa, A.; Stabile, E.; Di Lorenzo, E.; Esposito, G.; Pisani, A.; Leccia, A.; Cavuto, L.; Stingone, A.M.; Chieffo, A.; et al. Effects of hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin on smooth muscle cell proliferation in vitro and neointimal formation in vivo after vascular injury. J. Am. Coll. Cardiol. 1999, 35, 214–221.
- Indolfi, C.; Di Lorenzo, E.; Perrino, C.; Stingone, A.M.; Curcio, A.; Torella, D.; Cittadini, A.; Cardone, L.; Coppola, C.; Cavuto, L.; et al. Hydroxymethylglutaryl coenzyme a reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation 2002, 106, 2118–2124.
- Pasqui, A.L.; Pastorelli, M.; Bova, G.; Di Renzo, M.; Leo, A.; Cercignani, M.; Palazzuoli, A.; Auteri, A.; Bruni, F.; Puccetti, L. Platelet hyperactivity after statin treatment discontinuation. Thromb. Haemost. 2003, 90, 476–482.
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Théroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397.
- Becher, T.; Schulze, T.J.; Schmitt, M.; Trinkmann, F.; El-Battrawy, I.; Akin, I.; Kälsch, T.; Borggrefe, M.; Stach, K. Ezetimibe inhibits platelet activation and uPAR expression on endothelial cells. Int. J. Cardiol. 2017, 227, 858–862.
- Hussein, O.; Minasian, L.; Itzkovich, Y.; Shestatski, K.; Solomon, L.; Zidan, J. Ezetimibe’s effect on platelet aggregation and LDL tendency to peroxidation in hypercholesterolaemia as monotherapy or in addition to simvastatin. Br. J. Clin. Pharmacol. 2008, 65, 637–645.
- Piguet, P.F.; Vesin, C.; Donati, Y.; Tacchini-Cottier, F.; Belin, D.; Barazzone, C. Urokinase receptor (uPAR, CD87) is a platelet receptor important for kinetics and TNF-induced endothelial adhesion in mice. Circulation 1999, 99, 3315–3321.
- Abifadel, M.; Elbitar, S.; El Khoury, P.; Ghaleb, Y.; Chémaly, M.; Moussalli, M.-L.; Rabès, J.-P.; Varret, M.; Boileau, C. Living the PCSK9 adventure: From the identification of a new gene in familial hypercholesterolemia towards a potential new class of anticholesterol drugs. Curr. Atheroscler. Rep. 2014, 16, 439.
- Giugliano, R.P.; Pedersen, T.R.; Park, J.-G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971.
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107.
- Guedeney, P.; Giustino, G.; Sorrentino, S.; Claessen, B.E.; Camaj, A.; Kalkman, D.N.; Vogel, B.; Sartori, S.; De Rosa, S.; Baber, U.; et al. Efficacy and safety of alirocumab and evolocumab: A systematic review and meta-analysis of randomized controlled trials. Eur. Heart J. 2019, 43, e17–e25.
- Guedeney, P.; Sorrentino, S.; Giustino, G.; Chapelle, C.; Laporte, S.; Claessen, B.E.; Ollier, E.; Camaj, A.; Kalkman, D.N.; Vogel, B.; et al. Indirect comparison of the efficacy and safety of alirocumab and evolocumab: A systematic review and network meta-analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 7, 225–235.
- Sundararaman, S.S.; Döring, Y.; van der Vorst, E.P.C. PCSK9: A Multi-Faceted Protein That Is Involved in Cardiovascular Biology. Biomedicines 2021, 9, 793.
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649.
- Wang, S.; Fu, D.; Liu, H.; Peng, D. Independent association of PCSK9 with platelet reactivity in subjects without statin or antiplatelet agents. Front. Cardiovasc. Med. 2022, 9, 934914.
- Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193.
- Gareri, C.; Polimeni, A.; Giordano, S.; Tammè, L.; Curcio, A.; Indolfi, C. Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias. J. Clin. Med. 2022, 11, 3884.
- Landmesser, U.; Haghikia, A.; Leiter, L.A.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Stoekenbroek, R.; Kastelein, J.J.P.; Ray, K.K. Effect of inclisiran, the small-interfering RNA against proprotein convertase subtilisin/kexin type 9, on platelets, immune cells, and immunological biomarkers: A pre-specified analysis from ORION-1. Cardiovasc. Res. 2020, 117, 284–291.
- Bartlett, J.; Predazzi, I.M.; Williams, S.M.; Bush, W.S.; Kim, Y.; Havas, S.; Toth, P.P.; Fazio, S.; Miller, M. Is Isolated Low High-Density Lipoprotein Cholesterol a Cardiovascular Disease Risk Factor? Circ. Cardiovasc. Qual. Outcomes 2016, 9, 206–212.
- Arca, M.; Veronesi, C.; D’erasmo, L.; Borghi, C.; Colivicchi, F.; De Ferrari, G.M.; Desideri, G.; Pontremoli, R.; Temporelli, P.L.; Perrone, V.; et al. Association of Hypertriglyceridemia with All-Cause Mortality and Atherosclerotic Cardiovascular Events in a Low-Risk Italian Population: The TG-REAL Retrospective Cohort Analysis. J. Am. Heart Assoc. 2020, 9, e015801.
- Van Cutsem, E.; Taieb, J.; Yaeger, R.; Yoshino, T.; Grothey, A.; Maiello, E.; Elez, E.; Dekervel, J.; Ross, P.; Ruiz-Casado, A.; et al. ANCHOR CRC: Results from a Single-Arm, Phase II Study of Encorafenib Plus Binimetinib and Cetuximab in Previously Untreated BRAFV600E-Mutant Metastatic Colorectal Cancer. J. Clin. Oncol. 2023, 41, 2628–2637.
- Bays, H.E.; Ballantyne, C.M.; Kastelein, J.J.; Isaacsohn, J.L.; Braeckman, R.A.; Soni, P.N. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension trial). Am. J. Cardiol. 2011, 108, 682–690.
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098.
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.P.; Koenig, W.; McGuire, D.K.; et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk. JAMA 2020, 324, 2268–2280.
This entry is offline, you can click here to edit this entry!