Red cell diseases encompass a group of inherited or acquired erythrocyte disorders that affect the structure, function, or production of red blood cells (RBCs). These disorders can lead to various clinical manifestations, including anemia, hemolysis, inflammation, and impaired oxygen-carrying capacity. Oxidative stress, characterized by an imbalance between the production of reactive oxygen species (ROS) and the antioxidant defense mechanisms, plays a significant role in the pathophysiology of red cell diseases.
- erythrocyte
- reactive oxygen species
- antioxidant
- oxidative stress
- sickle cell disease
- glucose 6-phosphate dehydrogenase deficiency
- Pyruvate Kinase Deficiency
1. Oxidative stress in healthy red blood cell
Red blood cells (RBCs) are exposed to endogenous and exogenous oxidants, commonly referred to as reactive oxygen species (ROS) and reactive nitrogen species (RNS). These oxidants comprise a large group of molecules with different properties, including cellular sources and preferred molecular targets, which and have been discussed in more detail elsewhere [1][2].
One of the main mechanisms of endogenous oxidant production involves oxyhemoglobin (HbO2). The autoxidation of HbO2 occurs spontaneously at a low rate, to yield superoxide (O2•–) and methemoglobin (Hb-FIII, MetHb) [3]. Superoxide itself is a weak oxidant, but can further react to make stronger oxidants. Superoxide can spontaneously dismutate to yield hydrogen peroxide (H2O2) and oxygen [4]. Hydrogen peroxide is a stronger oxidant that will react with thiols and metal centers [5]. Hydrogen peroxide reacts with HbO2 to yield ferryl hemoglobin, which can oxidize other proteins and lipids [6]. Furthermore, in the presence of one electron reductants such as FeII, H2O2 can also generate hydroxyl radical (HO•), one of the strongest biological oxidants [4]. Hydroxyl radical will react with most organic molecules at diffusion-controlled rates, to yield organic radicals that can propagate the oxidative damage [6].
RBCs will also be exposed to oxidants derived from endothelial and immune system cells, which generate nitric oxide (NO•), superoxide, peroxynitrite (ONOO-), H2O2 and hypochlorous acid (HOCl). Nitric oxide is produced by the endothelial enzyme nitric oxide synthase (NOS3) as a signal molecule to induce vasodilation, and by immune system cells at larger amounts by inducible NOS2, that result in the formation of more potent oxidizing species that can kill invading pathogens [7].
Proteins and lipids can be damaged by oxidants in RBCs. The ultimate result of oxidative damage to RBCs is hemolysis, the loss of membrane integrity and the release of hemoglobin and the other intracellular proteins. Free hemoglobin is particularly toxic and this is evident in several RBCs diseases [8].
The membrane of RBCs is composed of phospholipids, cholesterol, glycolipids and proteins (some of them glycosylated). The polyunsaturated fatty acids (PUFA), 18% of the total fatty acids in RBC [9], are the lipid components that are more susceptible to oxidation, in a series of reactions that trigger lipoperoxidation. The first event is the abstraction of a bis-allylic hydrogen from a polyunsaturated fatty acid, to yield a lipid derived radical, which rapidly reacts with molecular oxygen to yield a lipid-derived peroxyl radical (LOO•). This LOO• can subsequently subtract a hydrogen from a neighboring PUFA, and the reaction propagates as a chain reaction [10]. The further oxidation of LOOH yields reactive aldehydes such as hydroxynonenal and malondialdehyde.
Although the RBCs are exposed to large amounts of oxidants, both from endogenous and exogenous sources, they are well prepared to resist. Robust antioxidant defenses allow normal RBCs to survive 120 days in circulation. The main defenses against oxidant damage are provided by different enzymatic systems, aided by low molecular weight antioxidant and electron-rich molecules. The antioxidant defenses ultimately rely on the reducing power of NADPH, obtained from the oxidation of glucose by the pentose phosphate pathway.
As developed above, RBCs oxidative stress strongly depends on the balance among pathophysiological mechanisms producing ROS and enzymatic and non-enzymatic antioxidant systems. In this second part of the research, researchers aim to explore several RBC diseases where this balance is strongly altered both by increased ROS production or by diminished antioxidant capacity. This selection of RBC diseases is not exhaustive.
2. Sickle Cell Disease
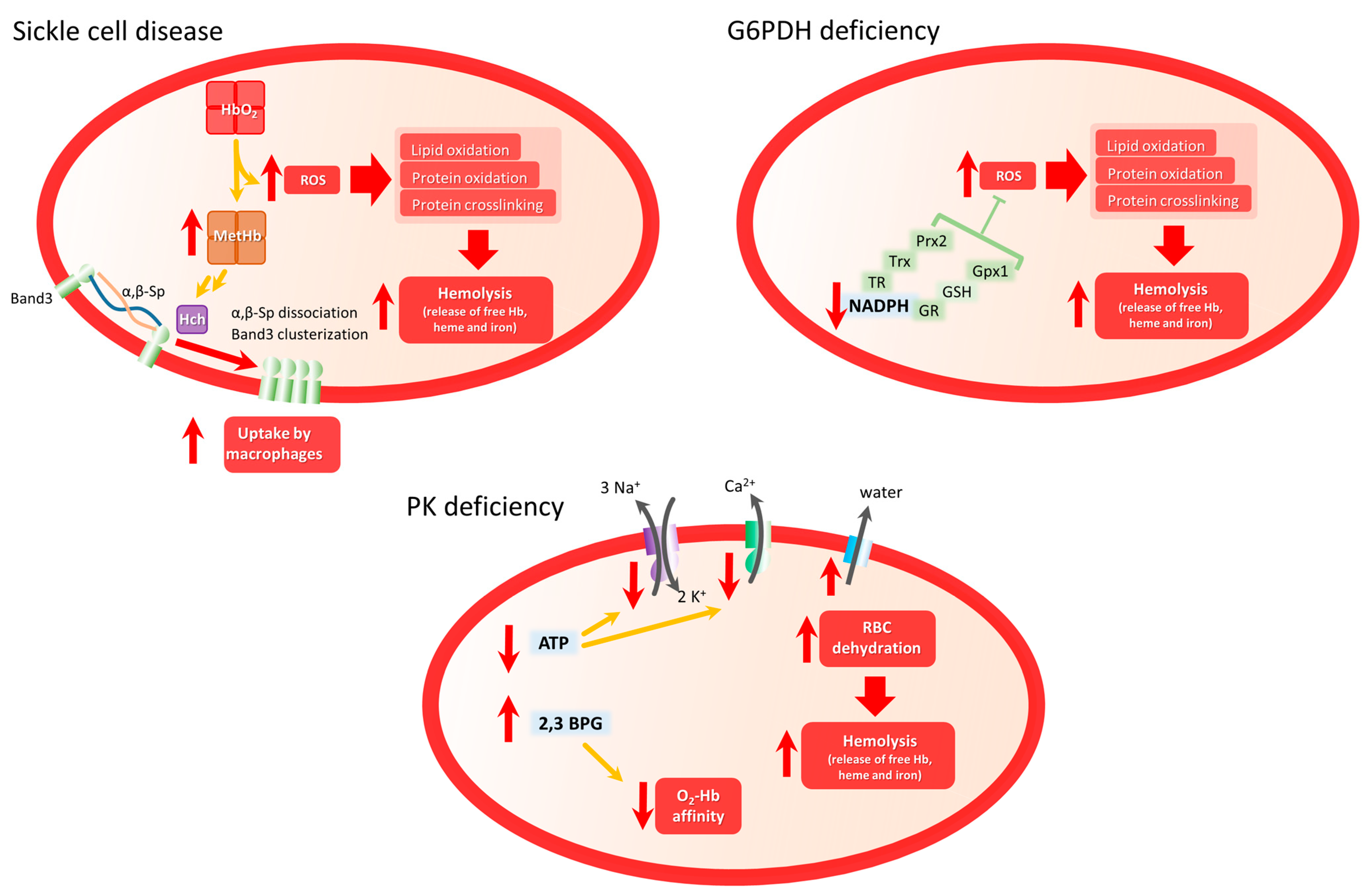
-
HbS autoxidation
-
Hemolysis: heme and iron release
-
NADPH oxidase and XO activity
-
NO• bioavailability
-
Erythroid mitochondrial retention
2. Glucose 6-Phosphate Dehydrogenase Deficiency
3. Pyruvate Kinase Deficiency
This entry is adapted from the peer-reviewed paper 10.3390/biom13081262
References
- Möller, M.N.; Cuevasanta, E.; Orrico, F.; Lopez, A.C.; Thomson, L.; Denicola, A. Diffusion and transport of reactive species across cell membranes. In Bioactive Lipids in Health and Disease; Trostchansky, A., Rubbo, H., Eds.; Springer: Cham, Switzerland, 2019; pp. 3–19.
- Moller, M.N.; Orrico, F.; Villar, S.F.; Lopez, A.C.; Silva, N.; Donze, M.; Thomson, L.; Denicola, A. Oxidants and Antioxidants in the Redox Biochemistry of Human Red Blood Cells. ACS Omega 2023, 8, 147–168.
- Johnson, R.M.; Goyette, G., Jr.; Ravindranath, Y.; Ho, Y.-S. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic. Biol. Med. 2005, 39, 1407–1417.
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543.
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 528, pp. 3–25.
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅O− in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886.
- Stuehr, D.J.; Haque, M.M. Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. Br. J. Pharmacol. 2019, 176, 177–188.
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284.
- Pacetti, D.; Gagliardi, R.; Balzano, M.; Frega, N.; Ojeda, M.; Borrero, M.; Ruiz, A.; Lucci, P. Changes in the fatty acid profile and phospholipid molecular species composition of human erythrocyte membranes after hybrid palm and extra virgin olive oil supplementation. J. Agric. Food Chem. 2016, 64, 5499–5507.
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972.
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296.
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241.
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84.
- Sheng, K.; Shariff, M.; Hebbel, R.P. Comparative oxidation of hemoglobins A and S. Blood 1998, 91, 3467–3470.
- Umbreit, J. Methemoglobin—It’s not just blue: A concise review. Am. J. Hematol. 2007, 82, 134–144.
- Welbourn, E.M.; Wilson, M.T.; Yusof, A.; Metodiev, M.V.; Cooper, C.E. The mechanism of formation, structure and physiological relevance of covalent hemoglobin attachment to the erythrocyte membrane. Free Radic. Biol. Med. 2017, 103, 95–106.
- Walder, J.A.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Arnone, A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. J. Biol. Chem. 1984, 259, 10238–10246.
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight 2018, 3, e120451.
- Camus, S.M.; Gausseres, B.; Bonnin, P.; Loufrani, L.; Grimaud, L.; Charue, D.; De Moraes, J.A.; Renard, J.M.; Tedgui, A.; Boulanger, C.M.; et al. Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood 2012, 120, 5050–5058.
- Tharaux, P.L. Posttranslational modifications of sickle hemoglobin in microparticles may promote injury. Kidney Int. 2019, 95, 1289–1291.
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814.
- Vinchi, F.; Sparla, R.; Passos, S.T.; Sharma, R.; Vance, S.Z.; Zreid, H.S.; Juaidi, H.; Manwani, D.; Yazdanbakhsh, K.; Nandi, V.; et al. Vasculo-toxic and pro-inflammatory action of unbound haemoglobin, haem and iron in transfusion-dependent patients with haemolytic anaemias. Br. J. Haematol. 2021, 193, 637–658.
- Woollard, K.J.; Sturgeon, S.; Chin-Dusting, J.P.; Salem, H.H.; Jackson, S.P. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J. Biol. Chem. 2009, 284, 13110–13118.
- Graca-Souza, A.V.; Arruda, M.A.; de Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil activation by heme: Implications for inflammatory processes. Blood 2002, 99, 4160–4165.
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827.
- Schimmel, M.; Nur, E.; Biemond, B.J.; van Mierlo, G.J.; Solati, S.; Brandjes, D.P.; Otten, H.M.; Schnog, J.J.; Zeerleder, S.; Curama Study, G. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 2013, 98, 1797–1803.
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107.
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 98, 15215–15220.
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 1995, 96, 60–68.
- Radomski, M.W.; Vallance, P.; Whitley, G.; Foxwell, N.; Moncada, S. Platelet adhesion to human vascular endothelium is modulated by constitutive and cytokine induced nitric oxide. Cardiovasc. Res. 1993, 27, 1380–1382.
- Simmonds, M.J.; Detterich, J.A.; Connes, P. Nitric oxide, vasodilation and the red blood cell. Biorheology 2014, 51, 121–134.
- Starzyk, D.; Korbut, R.; Gryglewski, R.J. The role of nitric oxide in regulation of deformability of red blood cells in acute phase of endotoxaemia in rats. J. Physiol. Pharmacol. 1997, 48, 731–735.
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624.
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 2017, 50, 46–52.
- Martino, S.; Arlet, J.B.; Odievre, M.H.; Jullien, V.; Moras, M.; Hattab, C.; Lefebvre, T.; Gouya, L.; Ostuni, M.A.; Lefevre, S.D.; et al. Deficient mitophagy pathways in sickle cell disease. Br. J. Haematol. 2021, 193, 988–993.
- Esperti, S.; Nader, E.; Stier, A.; Boisson, C.; Carin, R.; Marano, M.; Robert, M.; Martin, M.; Horand, F.; Cibiel, A.; et al. Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency, hemolysis and oxidative stress. Haematologica, 2023; Epub ahead of print.
- Moriconi, C.; Dzieciatkowska, M.; Roy, M.; D’Alessandro, A.; Roingeard, P.; Lee, J.Y.; Gibb, D.R.; Tredicine, M.; McGill, M.A.; Qiu, A.; et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br. J. Haematol. 2022, 198, 574–586.
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235.
- Cox, S.E.; Hart, E.; Kirkham, F.J.; Stotesbury, H. L-Glutamine in sickle cell disease. Drugs Today 2020, 56, 257–268.
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.; Stryer, L. Biochemistry, 9th ed.; W.H. Freeman/Macmillan Learning: New York, NY, USA, 2019; p. xlii.
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 371, 64–74.
- Luzzatto, L.; Ally, M.; Notaro, R. Glucose-6-phosphate dehydrogenase deficiency. Blood 2020, 136, 1225–1240.
- Beutler, E. G6PD deficiency. Blood 1994, 84, 3613–3636.
- Gomez Gomez, S.; Ruano Santiago, M.; Rodriguez Morillo, A.; Perez Munoz, A.M.; Echevarria Moreno, M. Anesthetic management of glucose 6-phosphate dehydrogenase deficiency. Rev. Esp. Anestesiol. Reanim. 2023, 70, 235–239.
- Wilson, J. Rasburicase-induced methaemoglobinaemia and catastrophic oxidative haemolysis in undiagnosed G6PD deficiency. Br. J. Haematol. 2023, 200, 7.
- Arese, P.; Mannuzzu, L.; Turrini, F. Pathophysiology of favism. Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. 1989, 116, 745–752.
- Luzzatto, L.; Arese, P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N. Engl. J. Med. 2018, 378, 60–71.
- Arese, P.; Bosia, A.; Naitana, A.; Gaetani, S.; D’Aquino, M.; Gaetani, G.F. Effect of divicine and isouramil on red cell metabolism in normal and G6PD-deficient (Mediterranean variant) subjects. Possible role in the genesis of favism. Prog. Clin. Biol. Res. 1981, 55, 725–746.
- McMillan, D.C.; Jollow, D.J. Favism: Divicine hemotoxicity in the rat. Toxicol. Sci. 1999, 51, 310–316.
- McMillan, D.C.; Schey, K.L.; Meier, G.P.; Jollow, D.J. Chemical analysis and hemolytic activity of the fava bean aglycon divicine. Chem. Res. Toxicol. 1993, 6, 439–444.
- Stamatoyannopoulos, G.; Fraser, G.R.; Motulsky, A.C.; Fessas, P.; Akrivakis, A.; Papayannopoulou, T. On the familial predisposition to favism. Am. J. Hum. Genet. 1966, 18, 253–263.
- Dinarelli, S.; Longo, G.; Germanova-Taneva, S.; Todinova, S.; Krumova, S.; Girasole, M. Surprising Structural and Functional Properties of Favism Erythrocytes Are Linked to Special Metabolic Regulation: A Cell Aging Study. Int. J. Mol. Sci. 2022, 24, 637.
- Francis, R.O.; D’Alessandro, A.; Eisenberger, A.; Soffing, M.; Yeh, R.; Coronel, E.; Sheikh, A.; Rapido, F.; La Carpia, F.; Reisz, J.A.; et al. Donor glucose-6-phosphate dehydrogenase deficiency decreases blood quality for transfusion. J. Clin. Invest. 2020, 130, 2270–2285.
- Pamuk, G.E.; Dogan Celik, A.; Uyanik, M.S. Brucellosis triggering hemolytic anemia in glucose-6-phosphate dehydrogenase deficiency. Med. Princ. Pr. 2009, 18, 329–331.
- Quereshy, F.A.; Gold, E.S.; Powers, M.P. Hemolytic anemia in a glucose-6-phosphate dehydrogenase-deficient patient triggered by a maxillofacial infection. J. Oral. Maxillofac. Surg. 2000, 58, 805–807.
- Araujo, T.; Katiyar, V.; Gonzales Zamora, J.A. Acute Retroviral Syndrome Presenting with Hemolytic Anemia Induced by G6PD Deficiency. Trop. Med. Infect. Dis. 2018, 4, 6.
- Meloni, T.; Forteleoni, G.; Porcu, A. Acute hemolytic anemia in two G6PD-deficient children with viral hepatitis. Haematologica 1988, 73, 397–399.
- Oluboyede, O.A.; Ayoola, E.A. Glucose 6 phosphate dehydrogenase enzyme (G6PD) and viral hepatitis in Nigeria. East. Afr. Med. J. 1982, 59, 754–759.
- Tang, H.Y.; Ho, H.Y.; Wu, P.R.; Chen, S.H.; Kuypers, F.A.; Cheng, M.L.; Chiu, D.T. Inability to maintain GSH pool in G6PD-deficient red cells causes futile AMPK activation and irreversible metabolic disturbance. Antioxid. Redox Signal 2015, 22, 744–759.
- Boonpeng, K.; Ketprasit, N.; Palasuwan, A.; Kulkeaw, K.; Palasuwan, D. Glucose-6-phosphate dehydrogenase is dispensable for human erythroid cell differentiation in vitro. Exp. Hematol. 2023, 121, 18–29.e2.
- Nelson, D.L.; Cox, M.M.; Lehninger, A.L. Lehninger Principles of Biochemistry, 7th ed.; W.H. Freeman and Company: New York, NY, USA; Macmillan Higher Education: Houndmills, UK, 2017; p. xxxiv.
- Bianchi, P.; Fermo, E.; Lezon-Geyda, K.; van Beers, E.J.; Morton, H.D.; Barcellini, W.; Glader, B.; Chonat, S.; Ravindranath, Y.; Newburger, P.E.; et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am. J. Hematol. 2020, 95, 472–482.
- Luke, N.; Hillier, K.; Al-Samkari, H.; Grace, R.F. Updates and advances in pyruvate kinase deficiency. Trends Mol. Med. 2023, 29, 406–418.
- Svidnicki, M.; Santos, A.; Fernandez, J.A.A.; Yokoyama, A.P.H.; Magalhaes, I.Q.; Pinheiro, V.R.P.; Brandalise, S.R.; Silveira, P.A.A.; Costa, F.F.; Saad, S.T.O. Novel mutations associated with pyruvate kinase deficiency in Brazil. Rev. Bras. Hematol. Hemoter. 2018, 40, 5–11.
- Zanella, A.; Fermo, E.; Bianchi, P.; Chiarelli, L.R.; Valentini, G. Pyruvate kinase deficiency: The genotype-phenotype association. Blood Rev. 2007, 21, 217–231.
- Chapman, R.G.; Schaumburg, L. Glycolysis and glycolytic enzyme activity of aging red cells in man. Changes in hexokinase, aldolase, glyceraldehyde-3-phosphate dehydrogenase, pyruvate kinase and glutamic-oxalacetic transaminase. Br. J. Haematol. 1967, 13, 665–678.
- Delivoria-Papadopoulos, M.; Oski, F.A.; Gottlieb, A.J. Oxygen-hemoglobulin dissociation curves: Effect of inherited enzyme defects of the red cell. Science 1969, 165, 601–602.
- van Wijk, R.; van Solinge, W.W. The energy-less red blood cell is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042.
- Bowman, H.S.; Oski, F.A. Splenic macrophage interaction with red cells in pyruvate kinase deficiency and hereditary spherocytosis. Vox Sang. 1970, 19, 168–175.
- Lakomek, M.; Winkler, H.; Pekrun, A.; Kruger, N.; Sander, M.; Huppke, P.; Schroter, W. Erythrocyte pyruvate kinase deficiency. The influence of physiologically important metabolites on the function of normal and defective enzymes. Enzym. Protein 1994, 48, 149–163.
- Oski, F.A.; Marshall, B.E.; Cohen, P.J.; Sugerman, H.J.; Miller, L.D. The role of the left-shifted or right-shifted oxygen-hemoglobin equilibrium curve. Ann. Intern. Med. 1971, 74, 44–46.
- Roy, M.K.; Cendali, F.; Ooyama, G.; Gamboni, F.; Morton, H.; D’Alessandro, A. Red Blood Cell Metabolism in Pyruvate Kinase Deficient Patients. Front. Physiol. 2021, 12, 735543.