Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pd-containing catalytic systems for tandem processes have gained special attention due to their enhanced catalytic properties and their possibility of performing several reactions without the necessity of separating the intermediates.
- tandem processes
- one-pot processes
- palladium
1. Introduction
Multiple catalytic processes in one reactor are of high importance due to the time–space economy and global tendency towards greener sustainable processes [1]. However, at present, there are many misinterpretations in the terminology used. As was mentioned by Camp et al. [2], such processes have different names: single-pot catalysis, one-pot catalysis, dominocatalysis, dual catalysis, tandem-catalysis, and multifaceted catalysis. Many papers use the term “tandem” for different processes proceeding one after another. Fogg and dos Santos [3] defined the one-pot catalytic processes that are not tandem catalysis as bicatalytic reactions, in which the second catalyst is added after the first one, which completes the reaction. When all the catalytic species are present in the reactor together, it can be defined as either domino or tandem catalysis. If all the reagents and catalysts are simultaneously present in the reactor, and the functionality formed in the previous step undergoes subsequent transformation, such a process can be classified as domino catalysis. It is important to emphasize that in many cases, domino catalysis is equated to the tandem catalysis, while Fogg and dos Santos [3] indentified of the domino/cascade catalysis as the multiple transformations proceeding via a single catalytic mechanism (Figure 1).
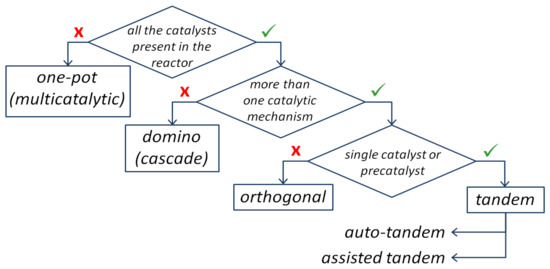
Figure 1. Distinguish between one-pot, domino, and tandem processes (based on [3]).
At present, homogeneous Pd-catalyzed processes, including the tandem ones, are the most widespread in fine organic synthesis for the production of active pharmaceutical ingredients (APIs) and polymers.
2. Coupling Tandem Processes
Palladium is known as a catalyst for the cross-coupling processes [4][5]. Both sequential and iterative Pd-catalyzed reactions can be carried out in tandem cross-coupling reactions, and the order of C–C bond forms can be regulated by either the attenuated leaving groups of the multireactive substrate or certain catalyst/ligand combinations [6][7][8][9]. Both the nucleophilic and electrophilic sites may be coupled in a specific manner. Additionally, in addition to halogen and metal leaving groups, the C–H bond can be used as an appealing leaving group [6].
In the catalytic cycle of cross-coupling reactions, palladium constantly changes its oxidation state from Pd(0) to Pd(II) and vice versa, hence the process can be catalyzed by any form of the metal [10][11]. Lamb et al. [12] employed Pd(II) to catalyze in the one-pot process the dehydrogenation of 2,2-disubstituted cyclopentane-1,3-diones and the oxidative Heck coupling. It was postulated that Pd(II) was an active form, which underwent partial decomposition during the reaction if the ligand loading was unsufficient. However, it was difficult to create the ideal one-pot conditions since the optimal conditions for the dehydrogenation stage were inappropriate for the oxidative Heck process.
By the example of Np-substituted cyclopentane-1,3-dione, it was shown that due to the presence of unligated Pd, which was likely formed during the aerobic dehydrogenation stage, the established one-pot reaction allowed for the achievement of a reasonable yield of 60% at moderate enantioselectivity at 74:26 e.r. In order to study the telescoped reaction, the reaction mixture was filtered to remove any unligated Pd before the cross-coupling partner was added. Such treatment enabled the e.r. to be comparable to the separate oxidative Heck step (88:12 vs. 90:10 e.r.) with agood 70% yield [12].
The improved synthesis of indolines via tandem decarboxylative amination/Heck/annulation reaction was carried out by Wang et al. [13]. Different palladium compounds (Pd(OAc)2, Pd(TFA)2,Pd2(dba)3, PdCl2) and ligands ((t-Bu)3P∙BF4, Ph3P, etc.) were used. PdCl2 and (t-Bu)3P∙BF4 revealed the best performance. Among the bases (Cs2CO3, K2CO3, t-BuOK, DBU, and Et3N), Cs2CO3 was optimum. Moreover, different solvents were used (benzene, toluene, xylene, MeCN, CHCl3, and DMF), and benzene was the most preferable one. Toluene was the second in efficiency allowing up to 70% yield of target product. In general, depending on the halogen, protecting group, substituent, and nature of the second substrate, as well as on the reaction conditions, up to 78% yield of the target product was achieved [13]. The reaction mechanism was proposed to proceed via formation of the intermediate of an intramolecular decarboxylation process. The cyclization product was then synthesized by reacting this intermediate with norbornadiene using a tandem Heck-type reaction and nitrogen nucleophilic route. The reaction followed a similar tandem Heck/intramolecular Tsuji–Trost pathway when the 1,3-diene substrate was used [13].
Song et al. [14] developed the synthesis of 2-(1-phenylvinyl)-indoles via the novel Pd(0)-catalyzed intermolecular coupling reaction of 2-gem-dibromovinylanilines and N-tosylhydrazones (Figure 2). Reaction conditions were optimized, (PdCl2(PPh3)2 was chosen as a catalyst, and tBuOLi was chosen as a base (the other bases were DMAP, tBuOK, TEA, CsCO3, and CsF)). Using 2-gem-dibromovinylanilines as a substrate, the scope of N-tosylhydrazones was studied. The reaction products were obtained in moderate to good yields (from 42% up to 94%), depending on the substituents: electrondonating (p-OMe and -OCH2O-) or electronwithdrawing (NO2 and CN). The substitutions on 2-gem-dibromovinylaniline were also studied (-CO2Me, Br, F, Ns, Ph, 4-F-Ph, 4-Cl-Ph, 4-OMe-Ph, 4-tBu-Ph, 4-Me-Ph, N-Ac, N-Ms), giving good yields (>80% in most of cases).
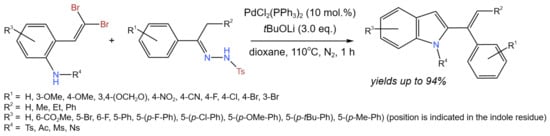
Figure 2. Scheme of the tandem reaction of 2-gem-dibromovinylanilines and N-tosylhydrazones for the synthesis of 2-(1-phenylvinyl)-indoles [14].
The reaction mechanism was also proposed, which included two catalytic cycles, one of which was started from Pd(0) and included oxidative addition, deprotonation, and reductive elimination, resulting in theformation of 2-bromo-1-tosyl-1H-indole. The latter entered the second cycle that also started from an oxidative addition to Pd(0) with the formation of the Pd(II)complex. Subsequently, alkylpalladium species were formed with carbene intermediate generated from the diazo compound. Finally, β-H elimination allowed for the synthesis of the desired product and the regeneration of Pd(0) [14].
In situ-produced vinyl fluorosulfate intermediates were coupled with electron-deficient olefin partners (Figure 3) by Revathi et al. [15] to create a protocol for the one-pot conversion of alcohols to 1,3-dienes. The use of DMSO as a solvent was mandatory, due to the involvement in the reaction mechanism. Broad substrate scopes of alcohols were examined for the SO2F2-mediated dehydrative cross-coupling. Different starting compounds containing both electron-donating groups (alkyl, aryl, trifluoromethoxy, methoxy, ether, thioether) and electron-withdrawing groups (nitro, trifluoromethyl) including halogens were investigated. Corresponding products were synthesized with the yields varying from 40% up to 99%. Moreover, the substrate scope of electron-deficient olefin coupling partners was studied. The best results were obtained with acrylamide, which provided the corresponding product at a 82% yield, and ethyl vinyl ketone, which produced the corresponding 1,3-diene at a 88% yield [15].
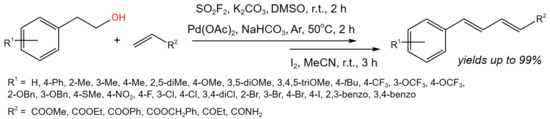
Figure 3. Scheme of dehydrative cross-coupling for the conversion of alcohols to 1,3-dienes [15].
The C–C/C–N bond formation in the reaction between N-methyl benzamide and arylboronic acid was catalyzed by the dinuclear palladium(II) complex, resulting in the direct synthesis of phenanthridinone (Figure 4) [16]. A broad range of phenanthridinones was synthesized in good to excellent yields (from 79% up to 96%) using 1 mol.% of the catalyst and a variety of substrates, which also included condensed aromatics and heterocycles (in the case of arylboronic acid).
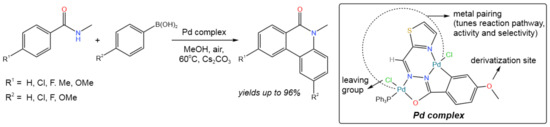
Figure 4. Scheme of the synthesis of phenanthridinones [16].
It was proposed that the reaction proceeds via ortho-arylation [16]. First, N-methyl arylamide was coordinated with the catalyst along with the ortho-C–H bond activation. Then, the transmetalation reaction occurred with participation of an arylboronic acid, leading to the formation of an ortho-arylated intermediate, which subsequently resulted in the seven-membered palladacycle. In this process, one of the two Pd(II) nuclei was transformed to Pd(0). Finally, the reductive elimination led to the desired product, while Pd(0) was oxidized by air.
The Pd pincer complex supported by 2,6-bis(pyrrolyl)pyridine ligands was shown [17] to be an efficient catalytic system for one-pot tandem Heck alkynylation (copper-free Sonogashira coupling)/cyclization reaction) (Figure 5). It was shown that a wide range of benzofuran derivatives can be obtained at 90 °C using 0.1 mol.% of catalyst loading for 10 h with high yields (up to 96%).
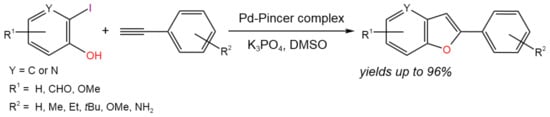
Figure 5. Scheme of the synthesis of benzofuran derivatives via the Heck alkynylation/cyclization reaction [17].
The reaction mechanism included two cycles (Heck alkynylation cycle and intramolecular cyclization cycle) associated with the product of Heck alkynylation, which underwent the cyclization process. It is noteworthy that the first cycle was likely initiated by Pd(II)/Pd(IV) transformations instead of the most common Pd(0)/Pd(II) chemistry [17].
In a similar way, microwave-assisted, domino [Pd]-catalyzed Heck cyclization followed by intermolecular Sonogashira coupling was carried out by Karu and Gedu [18] for the synthesis of substituted dihydrobenzofurans with yields of up to 99% (Figure 6). The reaction was proposed to proceed via a single catalytic cycle including classical Pd(0)/Pd(II) transformations. Initiated by Pd(0), the cyclization took place first, followed by Sonogashira coupling.
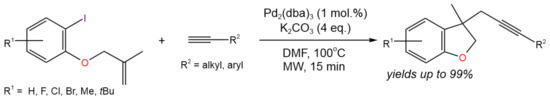
Figure 6. Scheme of the synthesis of substituted dihydrobenzofurans via Heck cyclization followed by Sonogashira coupling [18].
Such a kind of substituted heterocycles, containing a triple carbon–carbon bond, can be a subject of further transformations. For example, Ho et al. [19] developed the one-pot process for the dearomatising spirocyclization/cross-coupling of indole/pyrrole derivatives (Figure 7). In the proposed protocol the use of Pd(PPh3)4or trans-PdBr(N-succinimide)(PPh3)2 allowed up to 97% yields of target products. Palladium complexes were proposed to play a dual role: (i) π-acid to activating the alkyne towards dearomatising spirocyclization, and (ii) cross-coupling catalysts.
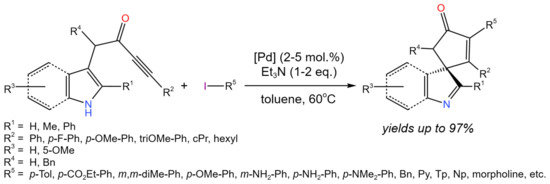
Figure 7. One-pot dearomatising spirocyclisation/cross-coupling [19].
Moreover, a variety of the spirocyclic pyrroline-based compounds, which are important scaffolds in drug development, could be produced with the yields up to 80% via Pd-catalyzed tandem Narasaka–Heck/C(sp3 or sp2)-H activation reaction [20] with γ,δ-unsaturated oxime esters as starting materials via the five-membered spiro-palladacycle intermediate.
Functionalized benzofurans, indoles, and phthalanes can be also synthesized by the tandem Ullmann–Goldberg cross-coupling and cyclopalladation-reductive elimination reactions, as well as by the related Pd-catalyzed processes involving hetero-Michael additions and cyclization in a one-pot process [21].
The tandem Heck/Suzuki reaction can be applied for the enantioselective intramolecular cyclization/cross-coupling of olefin-tethered aryl halides with various organoboronic acids [22]. Under the optimized reaction conditions, a number of dihydrobenzofuran derivatives were synthesized (Figure 8) with high yields (up to 99%) and e.r.s (up to 97:3). Moreover, by the same approach, different indolines, chromanes, and indanes were synthesized with yields of up to 99% and high e.r.s, depending on the substituents.
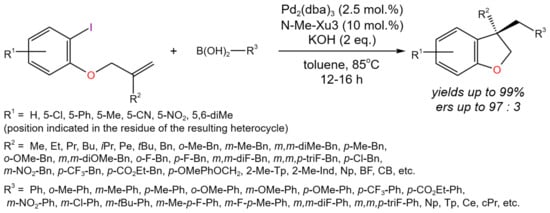
Figure 8. Scheme of the tandem Heck/Suzuki process for the synthesis of chiral compounds by the example of dihydrobenzofurans bearing a quaternary stereocenter [22].
Yokoya et al. [23] suggested fabricating the benzo[de]chromene ring followed by its oxidation in order to produce benzo[de]chromene-7,8-dione derivatives, which are known to have significant biological and pharmacological effects (Figure 9). Pd- and Cu-catalyzed Sonogashira coupling and cyclization was applied to produce benzo[de]chromene-7,8-dione derivatives with yields of up to 69%. It was found that the coupling process was catalyzed by Cu and Pd complexes, according to the established mechanism, while the further cyclization was catalyzed by CuI alone.
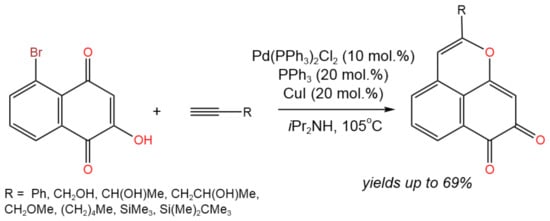
Figure 9. Tandem Sonogashira coupling and intramolecular cyclization with terminal alkynes for the synthesis of benzo[de]chromene-7,8-dione derivatives [23].
For the one-pot synthesis of alkenyl-substituted boron dipyrromethene (BODIPY) from Cl-BODIPY and alkyne, the coupling-reduction tandem process (Figure 10) was reported [24]. In comparison to traditional approaches, this synthesis enabled higher yields (up to 80%), a wider range of substrates, and a faster reaction rate by combining the Sonogashira coupling and the reduction process, avoiding the use of a reductant.
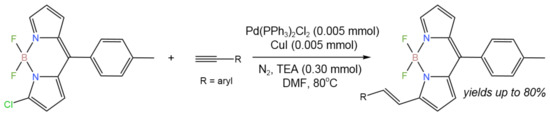
Figure 10. Synthesis of alkenyl-substituted BODIPYs [24].
One of the reactions attributed to the tandem Pd-catalyzed catalytic processes is the synthesis of imidazole derivatives [25][26][27]. The tandem synthesis of imidazole-fused polyheterocycles (Figure 11) from 2-vinyl imidazoles and aryl halides was reported by Li et al. [25] and involved intermolecular Heck arylation of 2-vinyl imidazoles, followed by an intramolecular aerobic oxidative C–H amination reaction.
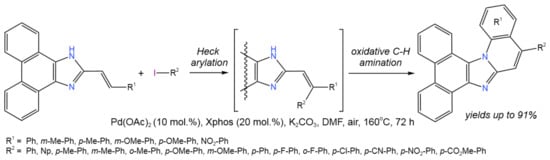
Figure 11. Pd-catalyzed annulation of phenanthroimidazoles with aryl iodides for the synthesis ring-fused phenanthroimidazoles [25].
In this tandem reaction, the Pd(0)↔Pd(II) catalytic cycle was used to simultaneously break two or three C–H bonds, one C–X bond, and one N–H bond. Different catalysts were used (Pd(OAc)2, PdCl2, Pd(PPh3)4, Pd2(dba)3), among which the palladium acetate allowed the highest yields of the target products (up to 91%). Potassium carbonate was chosen as an optimum base (other bases were Cs2CO3, NaOtBu, NaOH, and Et3N). DMF was shown to be the most preferable solvent as compared to DMA, DMSO, toluene, and 1,4-dioxane [25].
A tandem reaction involving boronic acids and 2-(bromobenzylsulfenyl)-1-propargyl benzimidazoles catalyzed by Pd was developed [26] (Figure 12). This process involved three different reactions: (i) Suzuki debrominative cross-coupling; (ii) Cu-free desulfenylative coupling at the 2-position of the benzimidazole; and (iii) intermolecular regioselective and stereoselective hydrothiolation of the triple C–C bond of the N-propargyl benzimidazole.
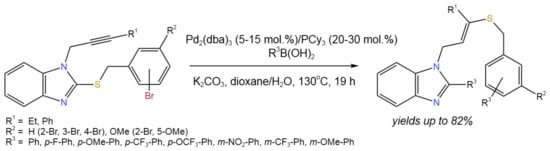
Figure 12. Tandem Pd-catalyzed reaction between boronic acids and 2-(bromobenzylsulfenyl)-1-propargyl benzimidazoles for the synthesis of substituted benzimidazoles bearing a stereodefined alkenyl sulfide [26].
The whole procedure resulted in an alkenyl benzyl sulfide and doubled the amount of boronic acid partner that was incorporated into the final structure. The developed tandem process proceeded with moderate up to high yields (82%), depending on the substituents and ligands. It is noteworthy that the heterocycles other than benzimidazole (i.e., imidazole and indole) were studied, and Pd(PPh3)4 (15 mol.%) was found to provide higher yields of target products as compared to Pd2(dba)3/PCy3 [26].
Luo et al. [27] developed the cross-coupling reaction of isocyanides with α-diazoacetates catalyzed by palladium to form ketenimines, which were subsequently underwent the DABCO-catalyzed aza-Mannich type reaction (Figure 13) under mild reaction conditions (80 °C, CH3CN—solvent). Different 2-mercaptoimidazoles or 1H-benzo[d]imidazol-2-oles were transformed to 1,3-bis(β-aminoacrylate)-substituted 2-mercaptoimidazoles and 2-benzimidazolinones with the yields up to 68%.
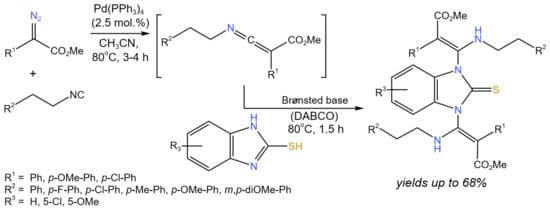
Figure 13. Scheme of the synthesis of 1,3-bis(β-aminoacrylate)-substituted 2-mercaptoimidazole derivatives synthesized in one pot using sequential Pd and Brønsted base catalysis [27].
The proposed reaction mechanism involved two cycles: (i) Pd-catalyzed, and (ii) DABCO-catalyzed. The starting isocyanide compound was coordinated with the palladium catalyst. Then, the diazo compound reacted with the formation of the Pd-carbene intermediate and the release of nitrogen. After that, the resulting intermediate produced ketenimine with regeneration of the catalyst. Ketenimine was trapped by one of the NH groups in DABCO-activated 2-mercaptoimidazole. The intermediate of this step reacted with ketenimine to form the target product [27]. Choi et al. [28] reported the tandem Diels–Alder/cross-coupling (either Stille or Suzuki) process using 2-bromo-1,3-butadienes as substrates, which underwent intermolecular cycloaddition with a variety of activated dienophiles in the presence of Lewis acids (e.g., BF3 · OEt2, TiCl4, SnCl4). The reaction proceeded with high yields (>70%) and good endo diastereoselectivity with respect to vinyl bromide cycloadducts, which further underwent cross-couplings in the presence of Pd catalysts (Pd2(dba)3, Pd(Ph3P)4).
3. Isomerization Tandem Processes
Isomerization tandem reactions catalyzed by Pd are possible due to the simultaneous existence of two oxidation states of this metal. Thus, there is double terminology for these reactions: some authors attribute them to orthogonal catalysis [29], othersto tandem catalysis [30]. Arroniz et al. [29] demonstrated the tandem orthogonal process of the conversion of alkynyl epoxides to furans (Figure 12). The developed procedure allowed for high yields (up to 93%) of furan derivatives.
It was found that the reaction proceeded through the formation of allenyl ketone and β,γ-alkynyl ketone as intermediates. The prposed reaction mechanism involved the oxidative addition of Pd(0) into the alkynylepoxide, resulting in the formation of allenylpalladium(II) species, which underwent reductive elimination to enol (shown in brackets in Figure 14). The latter tautomerized to alkynyl ketone and to allenyl ketone. The furanylpalladium complex was formed by the Pd(II)-activated cyclization of allenyl ketone. After the protodepalladation, the target furan’s derivatives were produced under mildly acidic conditions. Moreover, a possible alternative mechanism for the conversion of the alkyne to the furan was suggested [29].
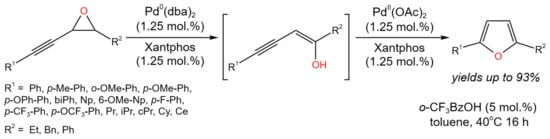
Figure 14. Dual catalytic isomerization of alkynyl epoxides for the synthesis of furans [29].
The synthesis of furans and pyrroles was carried out via the Pd-catalyzed tandem addition/ring-opening/cyclization reaction of 2-(3-aryloxiran-2-yl)acetonitriles with arylboronic acids in aqueous medium (Figure 15) [30].
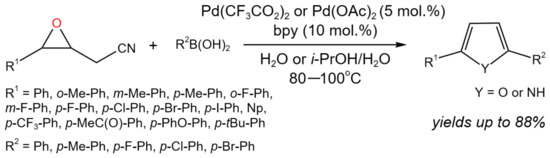
Figure 15. Tandem reaction of epoxyacetonitriles with arylboronic acids for the synthesis of furans and pyrroles [30].
Yu et al. [30] noticed that despite the advances in the Pd-catalyzed ring-opening of epoxides with nonpolar multiple bonds (such as vinyl and alkynyl), the reactions of boronic acids with epoxides with polar groups are less studied due to the lower reactivity.
As a result of the study, an efficient approach for the synthesis of furans and pyrroles with high selectivity was developed, allowing, depending on the catalyst and additives (p-toluenesulfonic acid, trifluoromethanesulfonic acid, D-camphorsulfonic acid, methanesulfonic acid, etc.), yields of up to 73% of furan derivatives (R1 = R2 = Ph) in aqueous medium at 80 °C under air. It is noteworthy that the replacement of air to the oxygen allowed for a noticeable increase in the yield of the target product (up to 86%). In the case of the synthesis of 2,5-diarylpyrroles at 100 °C under nitrogen using ai-PrOH/H2O mixture as a solvent, up to 84% yield was achieved, depending on the substituents. Moreover, it was found that the steric effects of arylboronic acids primarily controlled the chemo-selectivity of the developed tandem process: when 2-(3-aryloxiran-2-yl)acetonitriles were reacting with the less sterically hindering arylboronic acids, the only products were furans due to the hydrolysis of the ketimine intermediate, arising from the carbopalladation of the cyano group, whereas when reacting with the more sterically bulky arylboronic acids, pyrroles were selectively produced [30].
Another one example of isomerization tandem processes is the tandem isomerization/hydrothiolation of allylbenzenes (Figure 16) [31]. In the case of n = 0, using Pd(OAc)2 (3 mol.%) as a catalyst in the presence of 3.9 mol.% of the ligand and 12 mol.% of CF3SO3H, up to a 78% yield of the target product was able tobe achieved at 40 °C for 15 h, depending on the substituents. When n = 1–3 and R = H or 4-OMe, the one-pot process was carried out: (i) isomerization stage in the presence of PdCl2(PhCN)2 (5 mol.%) in toluene at 60 °C for 4 h; (ii) hydrothiolation stage in the presence of the ligand (10 mol.%), acid (22 mol.%), and thiol at 50 °C for 22 h, which allowed an up to 43% yield of the target product.
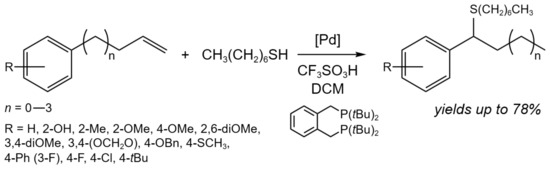
Figure 16. Tandem isomerization/hydrothiolation of allylarenes for the synthesis of benzylic thioethers [31].
It was demonstrated that the highly regioselective catalytic system composed of a Pd(II) precursor, bidentate phosphine ligand, and strong Brønsted acid may convert a wide range of 3-arylpropenes and thiols to branched benzylic thioethers. The reaction was facilitated by an in situ-generated Pd hydride [31].
4. Carbonylation Tandem Processes
Over 80% of all the APIs are known to have heterocyclic aromatic rings, with predominating N-containing aromatic heterocycles. Many APIs have the carbolinone ring system as a substructure [32]. Thus, carbonylation reactions are in high demand by the modern chemical industry. These reactions can be carried out in tandem with processes such as cross-coupling, dealkylation, and dehydrogenation.
A tetracyclic isoindoloindole skeleton with three new C–C/C–N bonds that was simultaneously generated, for instance, could be constructed in a one-step process using tandem Pd-catalyzed carbonylation and C–C cross-coupling via C–H activation (Figure 17) [33]. In more detail, the production of 6H-isoindolo[2,1-a]indol-6-ones from commercially accessible substrates involved the carbonylation of aryl dibromides with indoles and the C–H activation of in situ-produced N-(2′-bromoaroyl)-indole. C–H activation likely occurred via conventional Pd(0)/Pd(II) transformations. It is noteworthy that glyoxylic acid monohydrate was used as an eco-friendly CO surrogate in the aminocarbonylation stage of the proposed tandem process. As a result, a number of 6H-isoindolo[2,1-a]indol-6-ones were produced with yields of 40–64%.
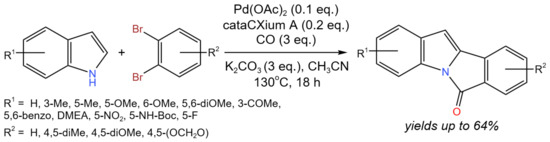
Figure 17. Tandem Pd-catalyzed carbonylation and C-C cross-coupling [33].
Li et al. [34] reported the enantioselective dearomative carbonylation of N-arylacyl indoles (Figure 18). The following reaction conditions were varied: Pd source (Pd2(dba)3·CHCl3, Pd(dba)2, Pd2(dba)3, Pd(OAc)2, Pd(TFA)2), chiral ligands, base additives (Et3N, DIPEA, DABCO, K2CO3), and solvents (MTBE, MeCN, THF, Et2O, PhOMe, toluene). Under the optimal conditions (Pd2(dba)3, Feringa ligand (S,S,S)-L1, Et3N, MTBE) under the atmosphere of CO, the target products were formed yields of up to 89%, up to 97% ee, and dr > 20:1.
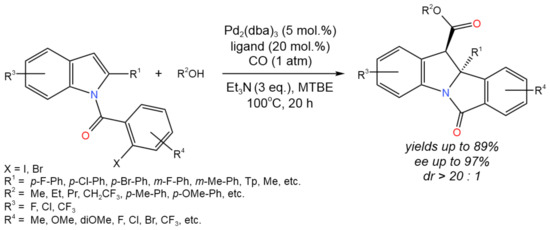
Figure 18. The scheme of thetandem Heck/carbonylation process for the asymmetric dearomatization of N-arylacyl indoles [34].
Wang et al. [35] developed the Pd-catalyzed multi-step tandem carbonylation/N-dealkylation/carbonylation reaction with alkyl as the leaving group and tertiary anilines as the nitrogen nucleophile, which allowed for the effective production of isatoic anhydride derivatives (Figure 19). Moderate to good (up to 80%) yields were obtained, depending on the substituents. Moreover, the developed approach was successfully used to synthesize Evodiamine—a biologically active alkaloid—with a 70% yield. The reaction mechanism involved two cycles. In the first cycle, the starting compound interacted with Pd(II) and CO. After the CO insertion and reductive elimination, the intermediate was formed, which then interacted with Cu(II) and O2 and underwent C-N cleavage. Then, the resulting intermediate entered the second cycle and underwent another one CO insertion with the participation of Pd(II). After the reductive elimination and formation of the target product, the Pd(0) was oxidized by Cu(II) and O2 to regenerate Pd(II).
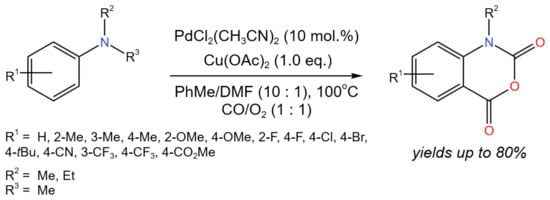
Figure 19. Tandem Pd-catalyzed carbonylation/N-dealkylation/carbonylation reaction of N,N-dialkyl anilines for the synthesis of isatoic anhydride derivatives [35].
For the synthesis of a variety of carbolinones under mild reaction conditions, Han et al. [32] proposed an effective tandem oxidative C–H aminocarbonylation and a dehydrogenation reaction co-catalyzed by Pd and Cu (Figure 20).
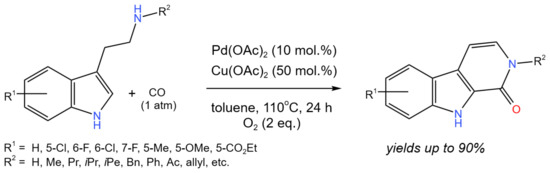
Figure 20. The scheme of the synthesis of β-carbolinones via the Pd/Cu co-catalyzed tandem C–H aminocarbonylation and dehydrogenation [32].
Carbolinones, tetrahydro-β-carbolinones, and tetrahydro-γ-carbolinoneswere selectively obtained with the yields of up to 90% by simply changing the reaction conditions. Strychnocarpine, a natural alkaloid, and its analogs were also produced.
The reaction proceeded through the well-known Pd(0)/Pd(II) transformations. The role of Cu(II) was in the oxidation of Pd(0) to regenerate Pd(II). In this process, Cu(II) was reduced to Cu(I), which was reoxidized with O2.
A new P,O-hybrid ligand (L1) was developed by Zhao et al. [36] for the Pd-catalyzed bis-hydroaminocarbonylation of alkynes, allowing for the effective production of N-aryl substituted succinimides with the isolated yield of 57–90% (Figure 21) in the presence of methanesulfonic acid (MSA) as an additive. The catalyst L1-Pd(CH3CN)2Cl2 system was reused for five runs without the precipitation of Pd-black.
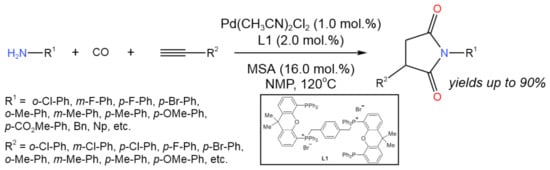
Figure 21. Pd-catalyzed bis-hydroaminocarbonylation of alkynes for the synthesis of N-aryl substituted succinimides [36].
One of the widespread reactions for the synthesis of cyclic compounds is the Diels–Alder reaction, which can be also combined with the carbonylation process [37]. Such an approach can provide a variety of functionalized carbocycles with complex architectures. Most of the methods of the synthesis of lactone-containing bridged polycyclic compounds are based on a stepwise protocol, restricting the overall scope and practicality of these transformations.
Therefore, the Pd-catalyzed tandem carbonylative Diels–Alder process was developed by Wang et al. [37] (Figure 22).
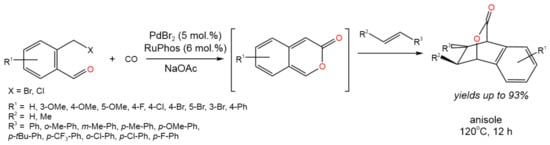
Figure 22. Scheme of the reaction of aldehyde-tethered benzylhalides and alkenes via the Pd-catalyzed tandem carbonylative lactonization and Diels–Alder cycloaddition [37].
It was proposed that the use of the aldehyde functionality as the reactive directing group resulted in good chemo- and stereoselectivity. Moderate to high (93%) yields were achieved, depending of the palladium source, base, and nature of substituents in the initial compounds [37]. The reaction was catalyzed by RuPhos-stabilized Pd(0), which was formed by the reduction of the Pd(II) precursor with CO or the ligand (RuPhos). Then, Pd(0) reacted with the substrate (benzaldehyde compound), producing an intermediate, which further underwent the insertion of CO. The subsequent reductive elimination resulted in the regeneration of Pd(0) and the formation of an intermediate, which underwent deprotonation due to interaction with NaOAc. Finally, the resulting benzopyran-2-one (shown in brackets in Figure 22) underwent [4 + 2]-cycloaddition with an alkene.
5. Cyclization Tandem Processes
Cyclization is another type of reaction widely used in fine synthesis [38][39][40][41][42][43][44][45][46][47]. Such reactions can proceed with the in situ-generated active species as a part of tandem catalytic transformations. For example, in order to produce EGFR (epidermal growth factor receptor) inhibitors, Ansari et al. [43][44] established a four-component procedure for the synthesis of pyrazolo[1,5-c]quinazolines with a small polar substitution of the pyrazole ring (Figure 23). In this tandem process, azomethine imine was generated in situ. Then, acetonitrile, having the electron-withdrawing group (EWG) at α-position (malononitrile, α-cyanocaboxyalate, orβ-ketonitrile), participated in the reaction.
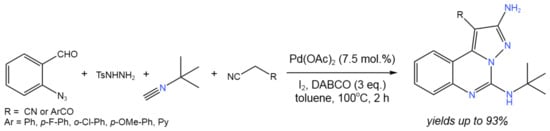
Figure 23. Four-component one-pot synthesis of pyrazolo[1,5-c]quinazoline [43].
In order to create 6-fluoroalkyl-phenanthridines in the absence of an oxidant, Bao et al. [48] developed a Pd-catalyzed tandem cyclization of fluorinated imidoyl chlorides using 2-bromophenylboronic acid (Figure 24). The developed procedure allowed for high yields (up to 97%) of target compounds using fluorinated imidoyl chlorides as fluorine-containing synthons. The authors [48] proposed the generally accepted Pd(0)/Pd(II) mechanism with Pd(0) as a starting active form, which interacts with fluorinated imidoyl chloride to form Pd(II). However, it was mentioned that the other pathway Pd(II)/Pd(IV) cannot be excluded.
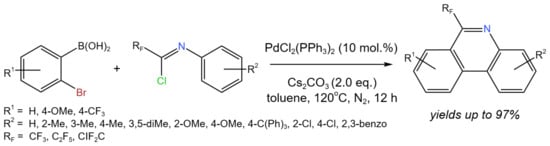
Figure 24. Synthesis of 6-fluoroalkyl-phenanthridines [48].
Another example of cyclization reactions proceeding with the participation of organic cyanides is the Pd-catalyzed tandem reaction of cyanomethyl benzoates with arylboronic acids developed by Dai et al. [49]. According to this approach, the selective synthesis of oxazoles and isocoumarins, through the intermediate imine–Pd complex obtained by carbopalladation of the nitrile, was found to depend strongly on substitution at the 2-position of starting cyano-compounds. Thus, 2,4-diaryloxazoles were selectively produced from cyanomethyl benzoates (3-benzoyl-4-aryl-isocoumarinsfrom 2-benzoyl-substituted cyanomethyl benzoates) with yields of up to 93% (Figure 25). To achieve high yields, different catalysts (Pd(acac)2, Pd(TFA)2, etc.), ligands (2,2’-bipyridine and its derivatives), additives (TfOH, TFA), and reaction conditions, such as solvent nature (THF, dioxane) and atmosphere (air, N2), were applied.
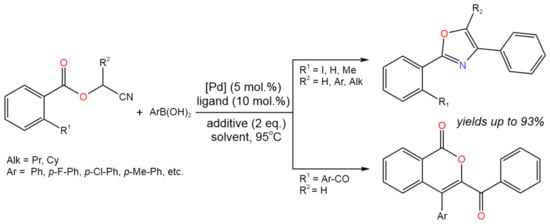
Figure 25. Scheme of the tandem reaction of cyanomethyl benzoates with arylboronic acids for the synthesis of oxazoles and isocoumarins [49].
By using the Pd-catalyzed tandem reaction of 2-aminostyryl nitriles with arylboronic acids, Xu et al. [50] developed a new protocol for the synthesis of 2-arylquinolines (Figure 26). The proposed procedure is an alternative synthetic route as compared to the common condensation reaction of (E)-2-aminostyryl ketones. It is noteworthy that the reaction was catalyzed by Pd(II), which interacted with arylbotonic acid, the resulting aryl-palladium complex, then reacted with the nitrile.
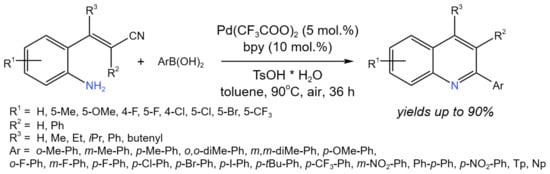
Figure 26. Pd-catalyzed tandem reaction of 2-aminostyryl nitriles with ArB(OH)2 [50].
In a similar manner, Yao etal. [51] implemented the tandem Pd-catalyzed cascade carbopalladation/cyclization/aromatization of arylboronic acids with 5-oxohexanenitrile and its derivatives to synthesize 2-methyl-6-arylpyridines. Different catalysts (Pd(OAc)2, Pd(CH3CN2)Cl2, Pd(CF3CO2)2, PdCl2, Pd(PPh3)2Cl2, Pd2(dba)3), ligands, and solvents (THF, DMSO, DMF, 1,4-dioxane, toluene, acetone, MeOH, EtOH, nPrOH, iPrOH, nBuOH, 1-PeOH, H2O) were studied. As a result, the optimum conditions were found (Pd2(dba)3 (5 mol.%), 2,2′-bipyridine ligand (10 mol.%), MeOH), which allowed for up to 94% yields of the target product for 12 h, depending on the substituents in the initial substrate molecules, with CF3COOH as an additive at 90 °C in air.
Ye et al. [52] established a palladium-catalyzed approach for the tandem reaction of 2-(arylamino)benzonitrile with arylboronic acids in water to produce 9-arylacridine derivatives posessing estrogenic biological activity (Figure 27).
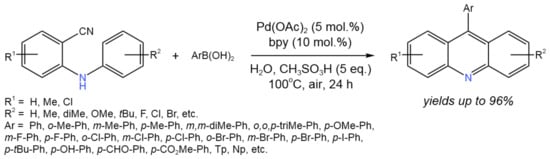
Figure 27. Tandem reaction of 2-(arylamino)benzonitrile with arylboronic acids for the synthesis of 9-arylacridine derivatives [52].
High yields of the desired products (up to 96%) were achieved via the proposed tandem process, involving the nucleophilic addition of aryl Pd species to the nitrile to produce an aryl ketone intermediate, which then underwent an intramolecular Friedel–Crafts acylation and dehydration to produce acridines.
A method for the production of benzoxazinones or quinazolinones was reported by Lang et al. (Figure 28) [53].
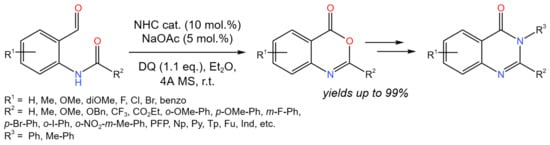
Figure 28. Carbene-catalyzed tandem isomerization/cyclisation for the synthesis of benzoxazinones or quinazolinones [53].
It was mentioned that other protocols are carried out under harsh conditions, restricting the functional group tolerance. According to the proposed strategy, benzoxazinone skeletons were synthesized via utilizing an oxidative carbene-catalyzed tandem isomerization/cyclisation process, which, under mild conditions, allowed for the production of a number of valuable benzoxazinones or quinazolinones from a wide range of substrates.
By using a one-pot intramolecular C–N coupling cyclization reaction, Patel et al. [54] created promising catalytic process to incorporate four-membered ring systems between the benzimidazole and 4-phenyl quinoline cores (Figure 29). This protocol was proposed to be an attractive route for the synthesis of different heterocycles, considering its high efficiency (yields up to 94%), wide substrate scope, and mild reaction conditions [54].
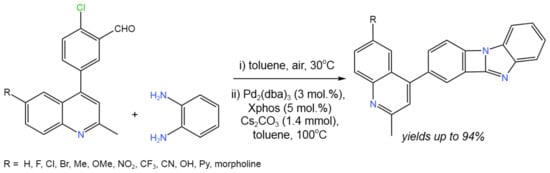
Figure 29. Scheme of the Pd-catalyzed intramolecular C–N coupling cyclization reaction [54].
The reaction likely begun with the condensation of o-phenylenediamine with 2-chloro-5-(N-substituted-2-methylquinoline-4-yl) benzaldehyde. The resulting intermediate then underwent C–N coupling via conventional Pd(II)/Pd(0) chemistry, starting with the oxidative addition to Pd(0) [54].
Another example of intramolecular cyclization is the work of Jin et al. [55], who developed new method for the synthesis of a cyclopenta-fused acenaphtho[1,2-b]indole (ANI) scaffold (Figure 30).
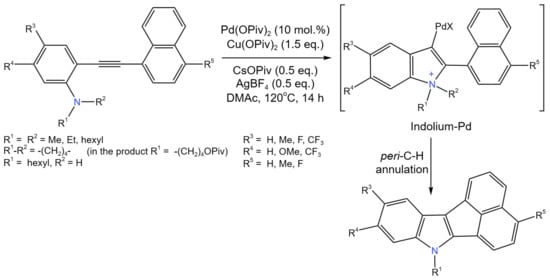
Figure 30. Scheme of Pd-catalyzed intramolecular C–N coupling cyclization reaction [55].
This Pd-catalyzed cascade process involved indolization, peri-C–H annulation, and N-dealkylation. Numerous o-alkynylanilines containing different substituents and tethered with various polyaromatic hydrocarbons having a peri-C–H bond were studied, allowing an up to 79% yield of the target product. It is noteworthy that, in contrast to the secondary aniline, the substrate with a N,N-dihexyl substituent resulted in an ANI with 100% selectivity [55].
In a similar way, Siciliano et al. [56] carried out the cyclisation of o-alkynyl-anilines using a ligand-free Pd(OAc)2 catalyst in 3 wt.% TPGS-750-M/H2O medium. Since the amino group was unprotected, the 2-substituted indoles were obtained with a yield of up to 76%, depending on the conditions (catalyst loading (2–10 mol.%), temperature (80–100 °C), type of heating (MW or oil bath), additive (AcOH, C11H23COOH, DPBA), and reaction duration (from 10 min to 16 h). Moreover, the tandem process Sonogashira/indole cyclisation was implemented according to the Cu-free mechanism, with the Pd(II) as the active species, which allowed for an up to 40% yield of the target product [56]. It is noteworthy that 2-substituted indoles can be also obtained via tandem Sonogashira coupling, followed by reductive cyclization with 1-halo-2-nitrobenzenes and terminal alkynes as starting compounds [40].
The Pd-catalyzed cascade addition/cyclization/aromatization of heteroarenes (e.g., thiophenes, furans, pyrroles, and indoles) with 2-(cyanomethoxy)chalcones was shown to be a promising approach for the synthesis of various benzofuro [2,3-c]pyridines (Figure 31) [57]. The following reaction conditions were varied: catalyst (PdCl2, Pd(CF3CO2)2, Pd(acac)2, Pd(OAc)2), solvent (DMA, NMA, THF, DMF, toluene), temperature (60–120 °C), oxidant (Ag2CO3, Cu(Oac)2, AgNO3, Ag2O, AgSbF6, AgCF3CO2), and additive (D-CSA, CF3CO2H, CF3SO3H, p-TsOH, CH3CO2H). As a result, the optimum parameters were chosen (catalyst Pd(OAc)2; solvent NMA; additive CH3CO2H; temperature ≥ 80 °C; and, optionally, AgCF3CO2), which provided the yields of target products of up to 86%.
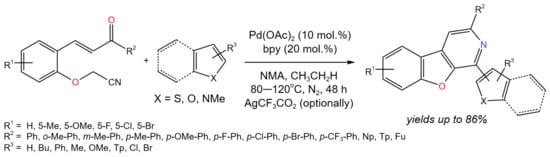
Figure 31. Pd-catalyzed cascade reaction of 2-(cyanomethoxy)chalcones with heteroarenes [57].
A novel procedure for the tandem intramolecular addition of active methylene compounds to internal alkynes, followed by coupling with aryl and heteroaryl bromides, was described by Błocka et al. [58] (Figure 32).
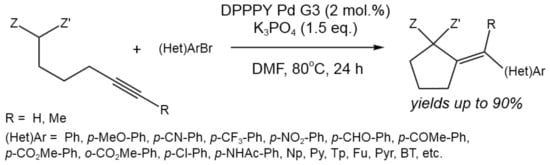
Figure 32. Tandem Pd-catalyzed carbocyclizationcoupling of methylene compounds bearing an internal alkyne group with aryl and heteroaryl bromides [58].
As a catalyst, third-generation (G3) Buchwald palladium precatalysts in combination with diphenyl-2-pyridylphosphine (DPPPY) ligand was used. This method allowed for the obtaining of a variety of vinylidenecyclopentanes with yields of up to 90%, providing high regio- and stereoselectivity and tolerance to the wide range of substituents. Thus, the the 5-exo-dig intramolecular addition proceeded with high efficiency.
The mechanism involving oxidative addition, cyclization, and reductive elimination was also confirmed by the computational study. The rate- and configuration-determining step was found to be the 5-exo-dig intramolecular nucleophilic addition of the enol intermediate to the alkyne activated via coordination with Pd(II) [58].
6. Other Tandem Processes
There are many other tandem reactions homogeneously catalyzed by palladium. This section will provide several examples, among which are alkylation, alkenylation arylation, cycloaddition, and carboannulation.
For example, Hu et al. [59] developed a one-pot approach for the synthesis of a triphenylene core under ligand-free conditionsusing commercially available (hetero)aromatic carboxylic acids and cyclic diaryliodonium salts (Figure 33). The obtained triphenylenes can be used as the electrontransport materials. The proposed reaction mechanism involved an acid-directed ortho-C–H arylation/intramolecular decarboxylative annulation sequence mediated by Pd(II)/Pd(IV) transformations.
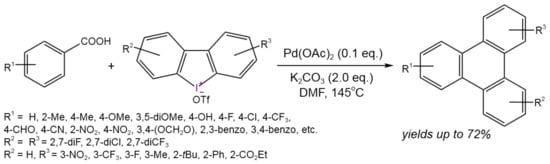
Figure 33. Scheme of tandem C–H arylation/decarboxylative annulation for the synthesis of functionalized triphenylenes [59].
The novel procedure of arylation-cyclization was developed for the synthesis of 3-aryl-2-quinolone (Figure 34) and 4-aryl-2-quinolone derivatives from simple E/Z geometric isomer precursors using oxygen as a co-oxidant [60].
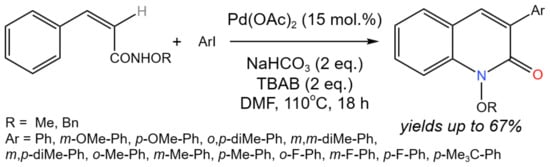
Figure 34. Scheme of tandem arylation-cyclization for the synthesis of 3-aryl-2-quinolones [60].
Ghosh and Chattopadhyay [45] identified the following Pd-catalyzed tandem procedures, which can be applied for the synthesis of 4-aryl-2-quinolones:
- (i)
-
intramolecular hydroarylation of yanamides;
- (ii)
-
hydroarylation-heterocyclization of 2-aminophenyl propiolate;
- (iii)
-
Heck-heterocyclization;
- (iv)
-
oxidative Heck-heterocyclization;
- (v)
-
arylation of ortho-halo-cinnamides, followed by Buchwald-type intramolecular amidation.
In the developed approach, the unusual complementary regioselectivity was found in the tandem C–H-arylation C–H-amidation of isomeric substrates. The origin of selectivity was proposed at the C–H-arylation step [60].
Domańskiet al. [61] described the tandem Pd-catalyzed three-component reaction that allowed for regio- and stereoselective perfluoroalkylative borylation of a variety of terminal and internal alkynes in the presence of perfluoroalkyl iodide and (Bpin)2. Thus, the first example of anti-addition across C–C multiple bonds of groups originating from two separate electrophiles to functionalize alkynes by reductive dicarbofunctionalization was shown. Iodoperfluoroalkylation, borylation, and coupling are the three fundamental processes responsible for this sequential transformation, and mechanistic studies have shown that their rates significantly differ (Figure 35).
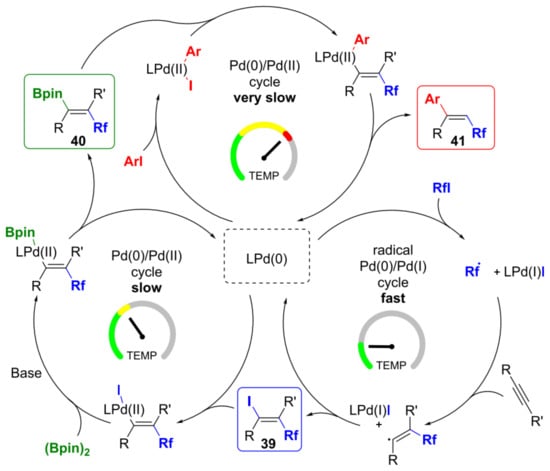
Figure 35. Proposed reaction mechanism of perfluoroalkylative borylation. Reproduced with permission from [61]. Copyright 2019 American Chemical Society.
According to the developed tandem process, from the same reaction mixture and under the same reaction conditions, the fluoroalkyl-substituted vinyl iodides, vinyl boronates, or olefins can be produced by changing the temperature program [61].
The Pd-catalyzed direct tandem C–O/C–H activation method for the C–C bond formation was described by Fernández et al. [62]. To accomplish base-free direct C–H alkenylation, the novel approach combined coordinated metalation–deprotonation of functionalized heterocycles with a C–O oxidative addition at enol pivalates (Figure 36).
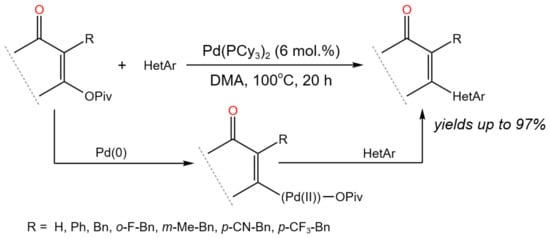
Figure 36. General scheme of Pd-catalyzed tandem C–O/C–H activation [62].
According to mechanistic studies, the C–O oxidative addition to Pd(0) was shown to be reversible, and the product of the Pd(II) C–O oxidative addition directly resulted in C–H activation [62].
Zhang et al. [63] developed a prospective route for the synthesis of poly-substituted quinolines via a three-component tandem reaction. 2-Aminobenzonitriles, arylboronic acids, and ketones were used as the substrates. Pd-catalyzed aryl addition to the cyano group was followed by hydrolysis and Friedländer-type cyclization to produce the quinoline compounds, with the yields of up to 96%. The described above cyclization tandem process developed by Xu et al. [39] resulted in lower yields, while comparing the same structures of target products.
Through the three-component tandem arylation and allylic etherification of 2,3-allenol with aryl iodides and alcohols, an effective approach for the synthesis of arylated allylic ethers was implemented [64]. By this method, functionalized 1-arylvinylated 1,2-diol derivatives were produced with yields of up to 83% and complete selectivities.
Tang et al. [65] reported a Pd-catalyzed tandem reaction of 3-allyloxybenzocyclobutenols that involved proximal C–C bond cleavage, C–O bond cleavage, and allylic alkylation of the C–H bond. Thus, a novel approach to meta-β-keto phenols bearing an allylic group with 100% atom economy was developed. It is noteworthy that, by adjusting substituents on various positions of benzocyclobutenols, sequential proximal or distal C–C bond cleavage/deallylation could be implemented.
Li et al. [66] developed a tandem process for the selective assembly of tri- or tetrasubstituted vinylsilanes. It was shown that using this method, ortho-vinyl bromobenzenes can be formed in situ from 1-bromo-2-iodobenzenes and N-tosylhydrazones and disilylated to produce two C–Si bonds and two C–C bonds.
It was determined that the ortho-vinyl bromobenzenes produced from 1-bromo-2-iodobenzenes and N-tosylhydrazones are the crucial intermediates needed to manufacture vinylsilanes by disilylating the C(aryl),C(vinyl)-palladacycle, which is formed by a direct vinylic C–H bond activation [66].
By the reaction with aldehydic N-tosylhydrazones, the treansformation of 1-aryl- and 2-aryl-1,2-dihydro-3H-indazol-3-ones into 1,2-di(hetero)aryl- and 2,3-di(hetero)aryl-2,3-dihydroquinazolin-4(1H)-ones was carried out (Figure 37) [67]. The method involved a cascade process that included base-mediated Pd-carbenoid production via the decomposition of N-tosylhydrazones, a nucleophilic attack of indazolone on the Pd-carbenoid complex, and intramolecular ring expansion by N–N bond cleavage.
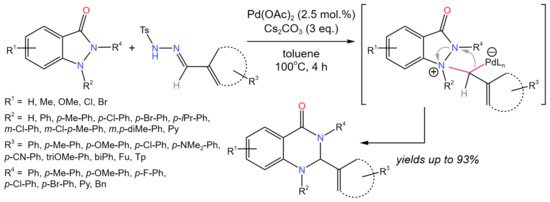
Figure 37. General scheme of Pd-catalyzed tandem C–O/C–H activation for the synthesis of 1,2-di(hetero)aryl- and 2,3-di(hetero)aryl-2,3-dihydroquinazolin-4(1H)-ones [67].
An interesting approach to the synthesis of [68][69]-spirocycles in a cascade Pd-catalyzed reaction was reported by Azizollahi et al. [70]. It was found that, when varying the type of ligand, the reaction pathway can be switched from carbopalladation to β-C-elimination: monodentate phosphine ligand (IMes·HCl, PCy3) affords [68][69]-spirocycles, while bidentate phosphines (Xantphos, DPPF, DPE-Phos) results in the formation of heteroaromatics.
Another example of switchable cascade reactions was the oxidation of N-allyl-2-aminophenols [71] in the presence of hypervalent iodines and Pd catalyst (Pd(OAc)2). In the absence of palladium, the dearomatization of the substrate and intramolecular Diels-Alder reaction occurred with the formation of tricyclic systems (yields of up to 72%), while the Pd-catalyzed process resulted in the methylacyloxylated dihydro-1,4-benzoxazines (yields of up to 86%).
Activated tetrasubstituted alkenes made from phthalides or butyrolactone were combined with vinylethylene carbonates in an unprecedented [5 + 2] cycloaddition/ring-contraction tandem process by Xiong et al. [72] under Pd(0) catalysis. The proposed approach was practical and mechanistically novel. In contrast to the traditional spirolactonization method, benzo-[69][69]-spiroketal lactones and [69][69]-spiroketal lactones bearing two vicinal tetrasubstituted centers can be obtained by this tandem [5 + 2] cycloaddition/ring-contraction mechanism with moderate to high yields.
It was proposed that the reaction was started from the formation of a zwitterionic π-allyl palladium intermediate via the interaction of vinylethylene carbonate with Pd(0), accompanied by the decarboxylation. Then, the intermediate reacted with activated phthalide or butyrolactone through [5 + 2] cycloaddition. Thus, a complex of Pd(0) and a spiro-oxepene phthalide was formed, which further facilitated the ring-opening reaction, resulting in a new zwitterionic π-allyl palladium intermediate. Finally, the branched allylic etherification occurred with the formation of a target product [72].
The first example of a transition-metal-catalyzed multicomponent carboannulation reaction of [60]fullerene was reported by Liu et al. [73]. In a Pd-catalyzed three-component tandem coupling–carboannulation process, [60]fullerene, 2-(2,3-allenyl)-malonates, and (hetero)aryl iodides were converted into a variety of polysubstituted [60]fullerene-fused cyclopentanes with a wide range of substrates and excellent functional group compatibility.
Different reaction conditions were explored: catalysts (Pd(PPh3)4, Pd(dba)2, Pd2(dba)3·· CHCl3), bases (Li2CO3, Cs2CO3, K2CO3, K3PO4, Rb2CO3, DMAP, and DABCO), and cosolvents (CH3CN, THF, 1,4-dioxane, DMF, and DMSO). Thus, the optimal conditions were found (Pd(PPh3)4, Rb2CO3, 100 °C, solvent 1,2-dichlorobenzene (ODCB), cosolvent CH3CN), which allowed for an up to 62% yield of the reaction product [73].
Zhang et al. [74] developed the Pd-catalyzed tandem synthesis of 2-trifluoromethylthio-(seleno)-substituted benzofurans, benzothiophenes, and indoles in acceptable to good yields (up to 93%). The reaction likely proceeded via the intramolecular cross-coupling followed by trifluoromethylthiolation.
Yang et al. [75] reported the asymmetric tandem C–C bond activation/Cacchi reaction between cyclobutanones and o-ethynylanilines. The chiral σ-alkylpalladium intermediates were formed via the enantioselective C(sp3)–C(sp2) bond activation of cyclobutanones and then promoted the cyclization of o-ethynylanilines, leading to one-carbon-tethered chiral indanone-substituted indoles. An all-carbon quaternary stereocenter was simultaneously formed, along with the two C–C bonds and one C–N bond. Under the optimum conditions (catalyst: [Pd(allyl)Cl]2 (0.05 eq.), TADDOL-derived phosphoramidite: L1 (0.1 eq.), base: K2CO3 (2.5 eq.), solvent: 1,4-dioxane, temperature: 90 °C), the indanone-substituted indoles with both central and axial stereogenic elements were synthesized with good yields (up to 85%) and excellent enantioselectivity (up to 99:1 e.r.).
The borrowing hydrogen (BH) reaction (hydrogen autotransfer) is another example of Pd-catalyzed tandem processes that can be mentioned [76] as a powerful strategy that combines transfer hydrogenation (avoiding the direct use of molecular hydrogen) with one or more intermediate reactions to synthesize more complex molecules without the need for time-consuming separation or isolation processes. The strategy of the BH process relies on three steps, namely, (i) dehydrogenation, (ii) intermediate reaction, and (iii) hydrogenation, among which the intermediate reaction works in tandem with the metal-catalyzed hydrogenation/dehydrogenation step.
This entry is adapted from the peer-reviewed paper 10.3390/catal13081213
References
- Campos, J.F.; Berteina-Raboin, S. Tandem Catalysis: Synthesis of Nitrogen-Containing Heterocycles. Catalysts 2020, 10, 631.
- Camp, J.E. Auto-Tandem Catalysis: Activation of Multiple, Mechanistically Distinct Process by a Single Catalyst. Eur. J. Org. Chem. 2017, 2017, 425–433.
- Fogg, D.E.; dos Santos, E.N. Tandem Catalysis: A Taxonomy and Illustrative Review. Coord. Chem. Rev. 2004, 248, 2365–2379.
- Balanta, A.; Godard, C.; Claver, C. Pd Nanoparticles for C–C Coupling Reactions. Chem. Soc. Rev. 2011, 40, 4973–4985.
- Biffis, A.; Centomo, P.; DelZotto, A.; Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295.
- Dobrounig, P.; Trobe, M.; Breinbauer, R. Sequential and Iterative Pd-catalyzed Cross-coupling Reactions in Organic Synthesis. Monatsh. Chem. 2017, 148, 3–35.
- Ueno, M.; Miyoshi, N.; Hanada, K.; Kobayashi, S. Three-Component, One-Pot Tandem Sonogashira/Suzuki-Miyaura Coupling Reactions for the Synthesis of a Library of Ceramide-Transport Protein Inhibitors Designed In Silico. Asian J. Org. Chem. 2020, 9, 267–273.
- Zhou, B.; Wang, H.; Cao, Z.-Y.; Zhu, J.-W.; Liang, R.-X.; Hong, X.; Jia, Y.-X. Dearomative 1,4-Difunctionalization of Naphthalenes via Palladium-Catalyzed Tandem Heck/Suzuki Coupling Reaction. Nat. Commun. 2020, 11, 4380.
- Matsude, A.; Hirano, K.; Miura, M. Highly Stereoselective Synthesis of 1,2-Disubstituted Indanes by Pd-Catalyzed Heck/Suzuki Sequence of Diarylmethyl Carbonates. Org. Lett. 2020, 22, 3190–3194.
- Pagliaro, M.; Pandarus, V.; Ciriminna, R.; Béland, F.; Carà, P.D. Heterogeneous versus Homogeneous Palladium Catalysts for Cross-Coupling Reactions. ChemCatChem 2012, 4, 432–445.
- Eremin, D.B.; Ananikov, V.P. Understanding Active Species in Catalytic Transformations: From Molecular Catalysis to Nanoparticles, Leaching, “Cocktails” of Catalysts and Dynamic Systems. Coord. Chem. Rev. 2017, 346, 2–19.
- Lamb, C.J.C.; Nderitu, B.G.; McMurdo, G.; Tobin, J.M.; Vilela, F.; Lee, A.-L. Auto-Tandem Catalysis: PdII-Catalysed Dehydrogenation/Oxidative Heck Reaction of Cyclopentane-1,3-diones. Chem. Eur. J. 2017, 23, 18282–18288.
- Wang, Z.; Li, P.; Fu, H.; Dai, Q.; Hu, C. Palladium-Catalyzed Synthesis of Indolines from Aroyloxycarbamates through a Tandem Decarboxylative Amination/Heck/Annulation Reaction. Adv. Synth. Catal. 2019, 361, 192–200.
- Song, J.; Chi, X.; Meng, L.; Zhao, P.; Sun, F.; Zhang, D.; Jiao, L.; Liu, Q.; Dong, Y.; Liu, H. Pd-Catalyzed Tandem Coupling Reaction of 2-gem-Dibromovinylanilines and N-Tosylhydrazones to Construct 2-(1-phenylvinyl)-indoles. Adv. Synth. Catal. 2019, 361, 3599–3604.
- Revathi, L.; Ravindar, L.; Balakrishna, M.; Qin, H.-L. SO2F2 mediated dehydrative cross-coupling of alcohols with electron-deficient olefins in DMSO using Pd-catalyst: One-pot transformation of alcohols to 1,3-diene. Org. Chem. Front. 2019, 6, 796–800.
- Manikandan, T.S.; Ramesh, R.; Semeril, D. The Tandem C–H/N–H Activation of N-Methyl Arylamide Catalyzed by Dinuclear Pd(II)Benzhydrazone Complex: A Concise Access to Phenanthridinone. Organometallics 2019, 38, 319–328.
- Yadav, S.; Dash, C. One-pot Tandem Heck Alkynylation/cyclization Reactions Catalyzed by Bis(Pyrrolyl)pyridine Based Palladium Pincer Complexes. Tetrahedron 2020, 76, 131350.
- Karu, R.; Gedu, S. Microwave Assisted Domino Heck Cyclization and Alkynylation: Synthesis of Alkyne Substituted Dihydrobenzofurans. Green Chem. 2018, 20, 369–374.
- Ho, H.E.; Stephens, T.C.; Payne, T.J.; O’Brien, P.; Taylor, R.J.K.; Unsworth, W.P. Merging π-Acid and Pd Catalysis: Dearomatizing Spirocyclization/Cross-Coupling Cascade Reactions of Alkyne-Tethered Aromatics. ACS Catal. 2019, 9, 504–510.
- Wei, W.-X.; Li, Y.; Wen, Y.-T.; Li, M.; Li, X.-S.; Wang, C.-T.; Liu, H.-C.; Xia, Y.; Zhang, B.-S.; Jiao, R.-Q.; et al. Experimental and Computational Studies of Palladium-Catalyzed Spirocyclization via a Narasaka–Heck/C(sp3 or sp2)–H Activation Cascade Reaction. JACS 2021, 143, 7868–7875.
- Khan, F.; Fatima, M.; Shirzaei, M.; Vo, Y.; Amarasiri, M.; Banwell, M.G.; Ma, C.; Ward, J.S.; Gardiner, M.G. Tandem Ullmann–Goldberg Cross-Coupling/Cyclopalladation-Reductive Elimination Reactions and Related Sequences Leading to Polyfunctionalized Benzofurans, Indoles, and Phthalanes. Org. Lett. 2019, 21, 6342–6346.
- Zhang, J.; Zhang, Z.-M.; Xu, B.; Wu, L.; Wu, Y.; Qian, Y.; Zhou, L.; Liu, Y. Enantioselective Dicarbofunctionalization of Unactivated Alkenes by Pd-Catalyzed Tandem Heck/Suzuki-Coupling Reaction. Angew. Chem. Int. Ed. 2019, 58, 14653–14659.
- Yokoya, M.; Ishiguro, T.; Sakairi, Y.; Kimura, S.; Morita, Y.; Yamanaka, M. Simple Strategy for Benzochromene-7,8-dione Synthesis via Tandem Sonogashira Coupling and Intramolecular Cyclization Reactions. Asian J. Org. Chem. 2022, 11, e202200534.
- Teng, K.-X.; Niu, L.-Y.; Li, J.; Jia, L.; Yang, Q.-Z. An Unexpected Coupling-Reduction Tandem Reaction for the Synthesis of Alkenyl-Substituted BODIPYs. Chem. Commun. 2019, 55, 13761–13764.
- Li, X.; Chen, X.; Wang, H.; Chen, C.; Sun, P.; Mo, B.; Peng, J. Palladium-Catalyzed Tandem One-pot Synthesis of π-Expanded Imidazoles through a Sequential Heck and Oxidative Amination Reaction. Org. Biomol. Chem. 2019, 17, 4014–4023.
- Lopes, A.B.; Choury, M.; Wagner, P.; Gulea, M. Tandem Double-Cross-Coupling/Hydrothiolation Reaction of 2-Sulfenyl Benzimidazoles with Boronic Acids. Org. Lett. 2019, 21, 5943–5947.
- Luo, J.; Chen, G.-S.; Chen, S.-J.; Li, Z.-D.; Zhao, Y.-L.; Liu, Y.-L. One-Pot Tandem Protocol for the Synthesis of 1,3-Bis(β-aminoacrylate)-Substituted 2-Mercaptoimidazole Scaffolds. Adv. Synth. Catal. 2020, 362, 3635–3643.
- Choi, H.; Shirley, H.J.; Aitken, H.R.M.; Schulte, T.; Söhnel, T.; Hume, P.A.; Brimble, M.A.; Furkert, D.P. Intermolecular Diels–Alder Cycloaddition/Cross-Coupling Sequences of 2-Bromo-1,3-butadienes. Org. Lett. 2020, 22, 1022–1027.
- Arroniz, C.; Chaubet, G.; Anderson, E.A. Dual Oxidation State Tandem Catalysis in the Palladium-Catalyzed Isomerization of Alkynyl Epoxides to Furans. ACS Catal. 2018, 8, 8290–8295.
- Yu, S.; Dai, L.; Shao, Y.; Li, R.; Chen, Z.; Lv, N.; Chen, J. Palladium-Catalyzed Tandem Reaction of Epoxynitriles with Arylboronic Acids in Aqueous Medium: Divergent Synthesis of Furans and Pyrroles. J. Org. Chem. Front. 2020, 7, 3439–3445.
- Kathe, P.M.; Fleischer, I. Palladium-Catalyzed Tandem Isomerization/Hydrothiolation of Allylarenes. Org. Lett. 2019, 21, 2213–2217.
- Han, H.; Yang, S.-D.; Xia, J.-B. Pd/Cu Cocatalyzed Oxidative Tandem C–H Aminocarbonylation and Dehydrogenation of Tryptamines: Synthesis of Carbolinones. J. Org. Chem. 2019, 84, 3357–3369.
- Čarný, T.; Markovič, M.; Gracza, T.; Koóš, P. One-Step Synthesis of Isoindoloindol-6-ones via Tandem Pd-Catalyzed Aminocarbonylation and C–HActivation. J. Org. Chem. 2019, 84, 12499–12507.
- Li, Q.; Zhang, Y.; Zeng, Y.; Fan, Y.; Lin, A.; Yao, H. Palladium-Catalyzed Asymmetric Dearomative Carbonylation of Indoles. Org. Lett. 2022, 24, 3033–3037.
- Wang, S.; Li, X.; Zang, J.; Liu, M.; Zhang, S.; Jiang, G.; Ji, F. Palladium-Catalyzed Multistep Tandem Carbonylation/N-Dealkylation/Carbonylation Reaction: Access to Isatoic Anhydrides. J. Org. Chem. 2020, 85, 2672–2679.
- Zhao, K.-C.; Zhuang, Y.-Y.; Jing, T.-H.; Shi, G.-H.; Guo, L.; Zhao, X.-L.; Lu, Y.; Liu, Y. Pd-Catalyzed Tandem Bis-Hydroaminocarbonylation of Terminal Alkynes for Synthesis of N-Aryl Substituted Succinimides with Involvement of Ionic P,O-hybrid Ligand. J. Catal. 2023, 417, 248–259.
- Wang, S.; Zhou, Y.; Huang, H. Palladium-Catalyzed Tandem Carbonylative Diels-Alder Reaction for Construction of Bridged Polycyclic Skeletons. Org. Lett. 2021, 23, 2125–2129.
- Cheng, Y.-J.; Zhao, L.-P.; Wang, L.; Tang, Y. Cyclization with Alkyl Substituted Methylene Malonate Enabling Concise Total Synthesis of Four Malagasy Alkaloids. CCS Chem. 2023, 5, 124–132.
- Gu, Z.-Y.; Xia, J.-B. Cyclization of vinyloxiranes with azides and CO by tandem palladium catalysis: Efficient synthesis of oxazolidinones. Org. Chem. Front. 2021, 8, 4112–4117.
- Lokolkar, M.S.; Mane, P.A.; Dey, S.; Bhanage, B.M. Synthesis of 2-Substituted Indoles by Pd-Catalyzed Reductive Cyclization of 1-Halo-2-nitrobenzene with Alkynes. Eur. J. Org. Chem. 2022, 2022, e202101505.
- Ding, L.; Niu, Y.-N.; Xia, X.-F. Pd-Catalyzed Tandem Isomerization/Cyclization for the Synthesis of Aromatic Oxazaheterocycles and Pyridoindoles. J. Org. Chem. 2021, 86, 10032–10042.
- Fillery, S.M.; Gregson, C.L.; Guérot, C.M. Expeditious Access to Functionalized Tricyclic Pyrrolo-Pyridones via Tandem or Sequential C–N/C–C Bond Formations. Org. Lett. 2019, 21, 9128–9132.
- Ansari, A.J.; Joshi, G.; Yadav, U.P.; Maurya, A.K.; Agnihotri, V.K.; Kalra, S.; Kumar, R.; Singh, S.; Sawant, D.M. Exploration of Pd-Catalysed Four-Component Tandem Reaction for One-Pot Assembly of Pyrazoloquinazolines as Potential EGFR Inhibitors. Bioorg. Chem. 2019, 93, 103314.
- Ansari, A.J.; Joshi, G.; Sharma, P.; Maurya, A.K.; Metre, R.K.; Agnihotri, V.K.; Chandaluri, C.G.; Kumar, R.; Singh, S.; Sawant, D.M. Pd-Catalyzed Four-Component Sequential Reaction Delivers a Modular Fluorophore Platform for Cell Imaging. J. Org. Chem. 2019, 84, 3817–3825.
- Gao, X.; Xia, M.; Yuan, C.; Zhou, L.; Sun, W.; Li, C.; Wu, B.; Zhu, D.; Zhang, C.; Zheng, B.; et al. EnantioselectiveSynthesis of Chiral Medium-Sized Cyclic Compounds via Tandem Cycloaddition/Cope Rearrangement Strategy. ACS Catal. 2019, 9, 1645–1654.
- Ren, Z.-L.; Qiu, J.-Y.; Yuan, L.-L.; Yuan, Y.-F.; Cai, S.; Li, J.; Kong, C.; He, P.; Wang, L. Divergent Conversion of Double Isocyanides with Alkenyl Bromide to Polysubstituted Pyrroles and 4-Imino-4,5-dihydropyrrolopyrrol-6(1H)-one Derivatives by Pd-Catalyzed Tandem Cyclization Reactions. Org. Lett. 2022, 24, 859–863.
- Wu, X.; Tang, Z.; Zhang, C.; Wang, C.; Wu, L.; Qu, J.; Chen, Y. Pd-Catalyzed Regiodivergent Synthesis of Diverse Oxindoles Enabled by the Versatile Heck Reaction of Carbamoyl Chlorides. Org. Lett. 2020, 22, 3915–3921.
- Bao, Y.; Wang, Z.; Chen, C.; Zhu, B.; Wang, Y.; Zhao, J.; Gong, J.; Han, M.; Liu, C. Palladium-Catalyzed Tandem Cyclization of Fluorinated Imidoyl Chlorides with 2-Bromophenylboronic Acid: Synthesis of 6-Fluoroalkyl-Phenanthridines. Tetrahedron 2019, 75, 1450–1456.
- Dai, L.; Yu, S.; Xiong, W.; Chen, Z.; Xu, T.; Shao, Y.; Chen, J. Divergent Palladium-Catalyzed Tandem Reaction of Cyanomethyl Benzoates with Arylboronic Acids: Synthesis of Oxazoles and Isocoumarins. Adv. Synth. Catal. 2020, 362, 1893–1898.
- Xu, T.; Shao, Y.; Dai, L.; Yu, S.; Cheng, T.; Chen, J. Pd-Catalyzed Tandem Reaction of 2-Aminostyryl Nitriles with Arylboronic Acids: Synthesis of 2-Arylquinolines. J. Org. Chem. 2019, 84, 13604–13614.
- Yao, X.; Qi, L.; Li, R.; Zhen, Q.; Liu, J.; Zhao, Z.; Shao, Y.; Hu, M.; Chen, J. Palladium-Catalyzed Cascade Reactions of δ-Ketonitriles with Arylboronic Acids: Synthesis of Pyridines. ACS Comb. Sci. 2020, 22, 114–119.
- Ye, X.; Xu, B.; Sun, J.; Dai, L.; Shao, Y.; Zhang, Y.; Chen, J. Pd-Catalyzed Approach for Assembling 9-Arylacridines via a Cascade Tandem Reaction of 2-(Arylamino)benzonitrile with Arylboronic Acidsin Water. J. Org. Chem. 2020, 85, 13004–13014.
- Lang, M.; Wang, J. Carbene-Catalyzed Tandem Isomerization/Cyclisation Strategy: Efficient Assembly of Benzoxazinones. Org. Chem. Front. 2019, 6, 1367–1371.
- Patel, J.J.; Patel, A.P.; Chikhalia, K.H. An Efficient Pd Catalyzed Intramolecular Cyclization Reaction Followed by Formation of Benzimidazole Derivatives: Synthesis of Novel Quinolin-Fused Benzo Azetobenzimidazole Analogues. Synth. Commun. 2021, 51, 81–93.
- Jin, T.; Suzuki, S.; Ho, H.E.; Matsuyama, H.; Kawata, M.; Terada, M. Pd-Catalyzed Indolization/peri-C–H Annulation/N-Dealkylation Cascade to Cyclopenta-Fused Acenaphthoindole Scaffold. Org. Lett. 2021, 23, 9431–9435.
- Siciliano, S.; Cini, E.; Taddei, M.; Vinciarelli, G. Synthesis of 2-Substitued Indoles via Pd-Catalysed Cyclization in an Aqueous Micellar Medium. Molecules 2021, 26, 3917.
- Xiong, W.; Chen, Z.; Shao, Y.; Li, R.; Hu, K.; Chen, J. The Synthesis of Fluorescent Benzofuro Pyridines via Palladium-Catalyzed Heteroaromatic C–H Addition and Sequential Tandem Cyclization. Org. Chem. Front. 2020, 7, 756–762.
- Błocka, A.; Chaładaj, W. Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides. Molecules 2022, 27, 630.
- Hu, T.; Xu, K.; Ye, Z.; Zhu, K.; Wu, Y.; Zhang, F. Two-in-One Strategy for the Pd(II)-Catalyzed Tandem C–H Arylation/Decarboxylative Annulation Involved with Cyclic Diaryliodonium Salts. Org. Lett. 2019, 21, 7233–7237.
- Ghosh, S.; Chattopadhyay, S.K. Unusual Regioselectivity in Palladium-Catalyzed Tandem C–H Arylation and C–H Amidation of cis-Cinnamyl Hydroxamates: Facile Synthesis of 3-Aryl-2-quinolones. Eur. J. Org. Chem. 2022, 2022, e202200391.
- Domański, S.; Gatlik, B.; Chaładaj, W. Pd-Catalyzed Boroperfluoroalkylation of Alkynes Opens a Route to One-Pot Reductive Carboperfluoroalkylation of Alkynes with Perfluoroalkyl and Aryl Iodides. Org. Lett. 2019, 21, 5021–5025.
- Fernández, N.P.; Gaube, G.; Woelk, K.J.; Burns, M.; Hruszkewycz, D.P.; Leitch, D.C. Palladium-Catalyzed Direct C–H Alkenylation with Enol Pivalates Proceeds via Reversible C–O Oxidative Addition to Pd(0). ACS Catal. 2022, 12, 6997–7003.
- Zhang, Y.; Chen, L.; Shao, Y.; Zhang, F.; Chen, Z.; Lv, N.; Chen, J.; Li, R. Palladium(ii)-catalyzed three-component tandem reactions: Synthesis of multiplysubstituted quinolines. Org. Chem. Front. 2021, 8, 254–259.
- Li, H.; Li, T.; Hsueh, Y.J.; Wu, X.; Xu, F.; Zhang, Y.J. Tandem arylation and regioselective allylic etherification of 2,3-allenol via Pd/B cooperative catalysis. Org. Biomol. Chem. 2019, 17, 8075–8078.
- Tang, T.-M.; Liu, M.; Wu, H.; Gou, T.; Hu, X.; Wang, B.-Q.; Hu, P.; Song, F.; Huang, G. Pd-Catalyzed tandem C–C/C–O/C–H single bond cleavage of 3-allyloxybenzocyclobutenols. Org. Chem. Front. 2021, 8, 3867–3875.
- Li, W.; Zhang, C.; Lu, H.; Wang, Y.; Deng, G.; Liang, Y.; Yang, Y. Pd-Catalyzed One-Pot Synthesis of Vinylsilanes via a Three-Component Tandem Reaction. Org. Chem. Front. 2020, 7, 2075–2081.
- Mahesha, C.K.; Borade, S.A.; Tank, D.; Bajaj, K.; Bhambri, H.; Mandal, S.K.; Sakhuja, R. Tandem Transformation of Indazolones to Quinazolinones through Pd-Catalyzed Carbene Insertionin to an N−N Bond. J. Org. Chem. 2023, 88, 1457–1468.
- Das, S.; Hong, D.; Chen, Z.; She, Z.; Hersh, W.H.; Subramaniam, G.; Chen, Y. Auto-Tandem Palladium Catalysis: From Isoxazoleto 2-Azafluorenone. Org. Lett. 2015, 17, 5578–5581.
- Chen, P.; Chen, Z.-C.; Li, Y.; Ouyang, Q.; Du, W.; Chen, Y.-C. Auto-Tandem Cooperative Catalysis Using Phosphine/Palladium: Reactionof Morita–Baylis–Hillman Carbonates and Allylic Alcohols. Angew. Chem. 2019, 131, 4076–4080.
- Azizollahi, H.; Mehta, V.P.; García-López, J.-A. Pd-catalyzed cascade reactions involving skipped dienes: From double carbopalladation to remote C–C cleavage. Chem.Commun. 2019, 55, 10281–10284.
- Giofrè, S.; Keller, M.; LoPresti, L.; Beccalli, E.M.; Molteni, L. Switchable Oxidative Reactions of N-allyl-2-Aminophenols: Palladium-Catalyzed Alkoxyacyloxylation vs an Intramolecular Diels–Alder Reaction. Org. Lett. 2021, 23, 7698–7702.
- Xiong, Q.; Lu, J.; Shi, L.; Ran, G.-Y. Pd-Catalyzed Tandem Cycloaddition/Ring Contraction of Phthalide-Derived Alkenes and Vinylethylene Carbonates for the Construction of Benzo--spiroketal Lactones. Org. Lett. 2022, 24, 3363–3367.
- Liu, Q.; Liu, T.-X.; Ma, J.; Zhang, G. Palladium-Catalyzed Three-Component Tandem Coupling–Carboannulation Reaction Leading to Polysubstituted Fullerene-Fused Cyclopentanes. Org. Lett. 2020, 22, 284–289.
- Zhang, M.; Weng, Z. Palladium-Catalyzed Tandem Synthesis of 2-Trifluoromethylthio(seleno)-Substituted Benzofused Heterocycles. Org. Lett. 2019, 21, 5838–5842.
- Yang, W.-C.; Chen, X.-B.; Song, K.-L.; Wu, B.; Gan, W.-E.; Zheng, Z.-J.; Cao, J.; Xu, L.-W. Pd-Catalyzed Enantioselective Tandem C–C Bond Activation/Cacchi Reaction between Cyclobutanones and o-Ethynylanilines. Org. Lett. 2021, 23, 1309–1314.
- Corma, A.; Navas, J.; Sabater, M.J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459.
This entry is offline, you can click here to edit this entry!