Extracellular vesicles (EVs) are complex phospholipidic structures actively released by cells. EVs are recognized as powerful means of intercellular communication since they contain many signaling molecules (including lipids, proteins, and nucleic acids).
- Extracellular vesicles in Epigenetic Regulation
1. Introduction
Currently, the intricate scenario of cell-to-cell communication is further complicated by the recognition of the pivotal role of EVs. EVs are complex phospholipidic cell-derived structures actively released from cells in the environment and able to shuttle biological information across cells and tissues. Although the involvement of EVs in virtually all biological processes (i.e., embryogenesis, neuronal plasticity, immune response) was demonstrated in the last decade, the existence of EVs was reported by Wolf in 1967, associated with platelets and coagulation [1], and they were subsequently observed in reticulocyte differentiation, acting as a cellular garbage system[2]. Since then, extensive research activity in the field has led to a better—although largely incomplete—understanding of their biological properties and has laid the foundation for their use as diagnostic and therapeutic tools[3]. Extracellular vesicles are classified into the following three main classes, accordingly to Minimum Information for Studies of EVs (MISEV) guidelines: small extracellular vesicles (sEVs, diameter < 200 nm, also known as exosomes), medium/large extracellular vesicles (m/lEVs, diameter >200 nm up to 1000 nm, also known as microvesicles), and apoptotic bodies (>1000 nm up to 5000 nm)[4]. Apart from the size, the three classes of EVs differ in their biogenesis[5] (Figure 1). Small EVs derive from the release of a larger structure, called a multivesicular body (MVB), a component of the endocytic pathway that sorts sEVs in its lumen, finally releasing them by the fusion with the plasma membrane. This mechanism involves multiple protein partners, such as Ras-related proteins in brain (RAB), endosomal sorting complex required for transport (ESCTR) components, and proteins in the ceramide/sphingomyelinase pathway[5]. Medium/large shedding EVs, or microvesicles, bud from the cell surface by sprouting and scission of the membrane. ADP-ribosylation factor 6 (ARF6) activates the phospholipase D (PLD), resulting in a phospholipidic reorganization and thereby relocating phosphatidylserines toward the outer side of the membrane. Finally, extracellular signal-regulated kinase (ERK) is recruited to the plasma membrane and activates through phosphorylation the myosin light-chain kinase (MLCK), resulting in invagination of plasma membrane and release of EVs[5]. Apoptotic bodies arise from the cleavage of the cell during apoptosis, a way to neatly package the cell components in the surroundings and exert many biological effects[6][7].
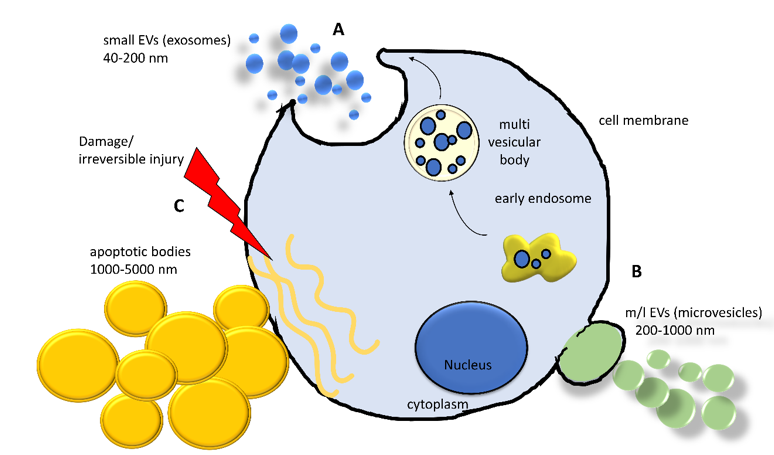
Figure 1. Schematization of the biogenesis and formation of the main classes of extracellular vesicles (EVs) released from a cell. (A) Early endosomes involved in the sorting and recycling of canonical intracellular vesicles can generate a multivesicular body (MVB), entrapping vesicles. An MVB can fuse with plasma membrane releasing its content in extracellular space. (B) Plasma membrane can undergo a complex remodeling by sophisticated molecular machinery, generating a bud from which a microvesicle is formed. (C) When a cell experiences a severe injury triggering irreversible damage, the cell activates the apoptotic pathway implying the organized dismantling of the cytoplasm. This process induces the release of the apoptotic bodies. sEVs = small extracellular vesicles; m/lEVs = medium/large extracellular vesicles.
Interestingly, all the resident bone cells, i.e., osteoblasts, monocytes/macrophages, osteoclasts, osteocytes, adipocytes, and endothelial cells, have been demonstrated to release EVs or respond to EVs both in physiological and pathological conditions[8][9][10][11][12][13][14][15]. EVs have been identified to shuttle molecules coming from the molecular legacy of the donor cells, exerting a specific effect according to the metabolic status of parental cells [16]. A multitude of studies defined the involvement of EVs in transferring genetic materials in many systems. For example, monocyte-derived EVs shuttle miR-155 to the endothelium, increasing endothelial cell migration[17]. Lv et al. demonstrated that renal tubular epithelial cells communicate by EVs shuttling miR-19b-3p to macrophages, leading to M1 macrophage switching[18]. Multiple myeloma cells under hypoxic conditions release miR-135b enriched sEVs able to decrease in target endothelial cells the factor-inhibiting hypoxia-inducible factor 1 (FIH-1), thereby increasing angiogenesis[19]. For a more detailed review on this aspect, see O’Brien et al.[20].
2. Control of Bone Metabolism by Means of EVs
Communication by means of EVs is a crucial mechanism involved in bone metabolism and intercellular crosstalk[21]. Some evidence in the late 1960s prompted researchers to postulate a role played by EVs in early mineral nucleation during cartilage mineralization[22][23]. More recently, Davies et al. demonstrated that mineralizing osteoblasts release EVs enriched in annexin 2, making the EV membrane able to complex octacalcium phosphate and other ions. This complex showed the intrinsic ability of triggering mineralization in an acellular context[24]. Furthermore, EVs are strictly related to cell-to-cell communication. Osteoclast differentiation and survival required the action of the irreplaceable cytokine receptor activator of NF-κB ligand (RANKL)[25][26][27]. Different groups described the release from osteoblast-like cells of EVs enriched in RANKL, directly supporting osteoclastogenesis[8][28]. In contrast, Ikebuchi et al. described that the mature osteoclasts produce EVs shuttling RANK[10]. Once bound to RANKL expressed on the osteoblast membrane, EVs trigger the reverse signaling pathway, inducing osteoblast maturation and bone deposition. Weilner and colleagues showed that endothelial cells produce EVs containing galectin-3, able to induce osteogenic differentiation on MSCs[29]. Adipocytes transfer via EVs adipocyte-specific transcripts such as adiponectin, resistin, and peroxisome proliferator-activated receptor gamma 2 (Pparγ2) into macrophages[30] or leptin, tumor necrosis factor alpha (Tnfα), and fibroblast growth factor alpha (Fgfα) into endothelial cells, thereby inducing angiogenesis[31]. Finally, peripheral blood mononuclear cells transfer via EVs the chemokine receptor CCR5 to endothelial cells[32]. Endothelial precursors have been described to secrete EVs able to attenuate steroid-induced osteoblast apoptosis and autophagy, being able to upregulate glutathione peroxidase 4, system Xc−, and cysteine levels while reducing malondialdehyde and reactive oxygen species production[33].
Here, we discuss in more detail some examples of miRNA and lncRNA transfer by EVs among the bone cells, focusing on mechanisms detrimental to bone quality (Figure 2).
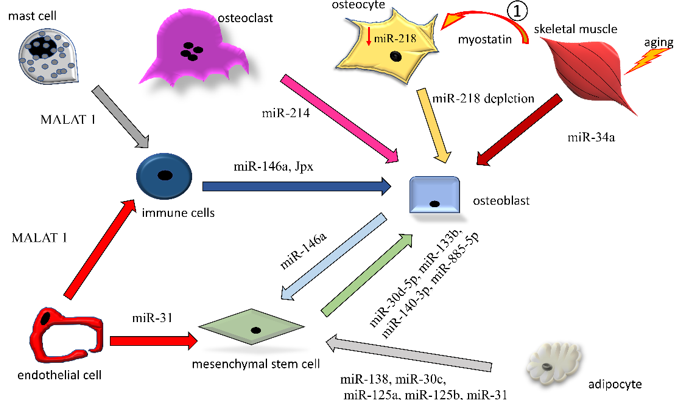
Figure 2. Intercellular exchanges of non-coding RNAs by means of EVs among bone-resident cells. Non-coding RNAs exerting osteopenic activity (increase in osteoclastogenesis or decrease in osteogenesis) are reported. Only ncRNAs directly found encompassed in EVs are reported in the figures. The arrows indicate the origin-to-destination direction between cells (donor to target cell).
1: soluble myostatin, released by aging muscle, affects osteocytes in a paracrine manner, reacting with the drop in endogenous miR-218a (↓miR-218a). The miR-218a-poor EVs released by the affected osteocytes are taken up by osteoblasts, contributing to the perturbation of osteogenesis.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21228682
References
- Shi, J.; Yu, J.; Zhang, Y.; Wu, L.; Dong, S.; Wu, L.; Wu, L.; Du, S.; Zhang, Y.; Ma, D. PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab. Invest. 2019, 99, 1795–1809.
- Yu, J.; Wang, Y.; Li, Z.; Dong, S.; Wang, D.; Gong, L.; Shi, J.; Zhang, Y.; Liu, D.; Mu, R. Effect of heme oxygenase-1 on mitofusin-1 protein in LPS-induced ALI/ARDS in rats. Sci. Rep. 2016, 6, 36530.
- Yu, J.; Shi, J.; Wang, D.; Dong, S.; Zhang, Y.; Wang, M.; Gong, L.; Fu, Q.; Liu, D. Heme oxygenase-1/carbon monoxide-regulated mitochondrial dynamic equilibrium contributes to the attenuation of endotoxin-induced acute lung injury in rats and in lipopolysaccharide-activated macrophages. Anesthesiology 2016, 125, 1190–1201.
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641-648.
- Suliman, H.B.; Carraway, M.S.; Ali, A.S.; Reynolds, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Invest. 2007, 117, 3730–3741.
- Suliman, H.B.; Carraway, M.S.; Tatro, L.G.; Piantadosi, C.A. A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 2007, 120, 299–308.
- Kim, H.J.; Joe, Y.; Rah, S.Y.; Kim, S.K.; Park, S.U.; Park, J.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; Ryter, S.W.; Kim, U.H.; Chung, H.T. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018, 9, 1060.
- Motterlini, R. Carbon monoxide-releasing molecules (CO-RMs): Vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem. Soc. Trans. 2007, 35 Pt 5, 1142–1146.
- Motterlini, R.; Sawle, P.; Hammad, J.; Bains, S.; Alberto, R.; Foresti, R.; Green, C.J. CORM-A1: A new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005, 19, 284–286.
- Fayad-Kobeissi, S.; Ratovonantenaina, J.; Dabiré, H.; Wilson, J.L.; Rodriguez, A.M.; Berdeaux, A.; Dubois-Randé, J.L.; Mann, B.E.; Motterlini, R.; Foresti, R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem. Pharmacol. 2016, 102, 64–77.
- Kretschmer, R.; Gessner, G.; Görls, H.; Heinemann, S.H.; Westerhausen, M. Dicarbonyl-bis(cysteamine)iron(II): A light induced carbon monoxide releasing molecule based on iron (CORM-S1). J. Inorg. Biochem. 2011, 105, 6–9.
- Wright, M.A.; Wright, J.A. PhotoCORMs: CO release moves into the visible. Dalton Trans. 2016, 45, 6801–6811.
- Mazzola, S.; Forni, M.; Albertini, M.; Bacci, M.L.; Zannoni, A.; Gentilini, F.; Lavitrano, M.; Bach, F.H.; Otterbein, L.E.; Clement, M.G. Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J. 2005, 19, 2045–2047.
- Mitchell, L.A.; Channell, M.M.; Royer, C.M.; Ryter, S.W.; Choi, A.M.; McDonald, J.D. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L891–L897.
- Fredenburgh, L.E.; Kraft, B.D.; Hess, D.R.; Harris, R.S.; Wolf, M.A.; Suliman, H.B.; Roggli, V.L.; Davies, J.D.; Winkler, T.; Stenzler, A.; Baron, R.M.; Thompson, B.T.; Choi, A.M.; Welty-Wolf, K.E.; Piantadosi, C.A. Effects of inhaled CO administration on acute lung injury in baboons with pneumococcal pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L834–L846.
- Kawanishi, S.; Takahashi, T.; Morimatsu, H.; Shimizu, H.; Omori, E.; Sato, K.; Matsumi, M.; Maeda, S.; Nakao, A.; Morita, K. Inhalation of carbon monoxide following resuscitation ameliorates hemorrhagic shock-induced lung injury. Mol. Med. Rep. 2013, 7, 3–10.
- Kanagawa, F.; Takahashi, T.; Inoue, K.; Shimizu, H.; Omori, E.; Morimatsu, H.; Maeda, S.; Katayama, H.; Nakao, A.; Morita, K. Protective effect of carbon monoxide inhalation on lung injury after hemorrhagic shock/resuscitation in rats. J. Trauma 2010, 69, 185–194.
- Kumada, Y.; Takahashi, T.; Shimizu, H.; et al. Therapeutic effect of carbon monoxide-releasing molecule-3 on acute lung injury after hemorrhagic shock and resuscitation. Exp. Ther. Med. 2019, 17, 3429–3440.
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Invest. 2008, 118, 239–247.
- Hoetzel, A.; Schmidt, R.; Vallbracht, S.; Goebel, U.; Dolinay, T.; Kim, H.P.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med. 2009, 37, 1708–1715.
- Morita, T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1786–1795.
- Hoetzel, A.; Dolinay, T.; Vallbracht, S.; Zhang, Y.; Kim, H.P.; Ifedigbo, E.; Alber, S.; Kaynar, A.M.; Schmidt, R.; Ryter, S.W.; Choi, A.M. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am. J. Respir. Crit. Care Med. 2008, 177, 1223–1232.
- Dolinay, T.; Szilasi, M.; Liu, M.; Choi, A.M. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 613–620.
- Song, R.; Kubo, M.; Morse, D.; Zhou, Z.; Zhang, X.; Dauber, J.H.; Fabisiak, J.; Alber, S.M.; Watkins, S.C.; Zuckerbraun, B.S.; Otterbein, L.E.; Ning, W.; Oury, T.D.; Lee, P.J.; McCurry, K.R.; Choi, A.M. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am. J. Pathol. 2003, 163, 231–242.
- Kohmoto, J.; Nakao, A.; Kaizu, T.; Tsung, A.; Ikeda, A.; Tomiyama, K.; Billiar, T.R.; Choi, A.M.; Murase, N.; McCurry, K.R. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 2006, 140, 179–185.
- Kohmoto, J.; Nakao, A.; Stolz, D.B.; Kaizu, T.; Tsung, A.; Ikeda, A.; Shimizu, H.; Takahashi, T.; Tomiyama, K.; Sugimoto, R.; Choi, A.M.; Billiar, T.R.; Murase, N.; McCurry, K.R. Carbon monoxide protects rat lung transplants from ischemia-reperfusion injury via a mechanism involving p38 MAPK pathway. Am. J. Transplant. 2007, 7, 2279–2290.
- Kohmoto, J.; Nakao, A.; Sugimoto, R.; Wang, Y.; Zhan, J.; Ueda, H.; McCurry, K.R. Carbon monoxide saturated preservation solution protects lung grafts from ischemia reperfusion injury. J. Thorac. Cardiovasc. Surg. 2008, 136, 1067–1075.
- Neto, J.S.; Nakao, A.; Toyokawa, H.; Nalesnik, M.A.; Romanosky, A.J.; Kimizuka, K.; Kaizu, T.; Hashimoto, N.; Azhipa, O.; Stolz, D.B.; Choi, A.M.; Murase, N. Low dose carbon monoxide inhalation prevents development of chronic allograft nephropathy. Am. J. Physiol. Renal Physiol. 2006, 290, F324–F334.
- Neto, J.S. Nakao, A.; Kimizuka, K.; Romanosky, A.J.; Stolz, D.B.; Uchiyama, T.; Nalesnik, M.A.; Otterbein, L.E.; Murase, N. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am. J. Physiol. Renal Physiol. 2004, 287, F979-F989.
- Nakao, A.; Faleo, G.; Nalesnik, M.A.; Seda-Neto, J.; Kohmoto, J.; Murase, N. Low-dose carbon monoxide inhibits progressive chronic allograft nephropathy and restores renal allograft function. Am. J. Physiol. Renal Physiol. 2009, 297, F19–F26.
- Nakao, A.; Toyoda, Y. Application of carbon monoxide for transplantation. Curr. Pharm. Biotechnol. 2012, 13, 827-836.
- Inoue, S.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Hashiba, T.; Tsuburai, T.; Ikehara, K.; Matsuse, T.; Ishigatsubo, Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum. Gene Ther. 2001, 12, 967–979.
- Hashiba, T.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Inoue, S.; Tsuburai, T.; Matsuse, T.; Ishigatubo, Y. Adenovirus-mediated transfer of heme oxygenase-1 cDNA attenuates severe lung injury induced by the influenza virus in mice. Gene Ther. 2001, 8, 1499–1507.