Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Genetically modified (GM) maize is one of the earliest GM crops to have achieved large-scale commercial cultivation globally, and it is of great significance to excel in the development and implementation of safety policy regarding GM, and in its technical oversight.The extraction technology of maize has an important impact on the quality of nucleic acid detection.The traditional nucleic acid detection technology used in maize covers variable-temperature amplification, isothermal amplification detection technology and gene chip technology.
- genetically modified maize
- amplification technology
- nucleic acid detection
1. Introduction
Maize is important in the food industry because of its nutritional characteristics and wide applications in the food industry. At the same time, maize is the food crop with the largest production volume (tons) in the world [1], and, thus, it is considered to be a strategic crop for national food security by many countries [2]. Therefore, the continuous demand for maize products has driven an increase in its production. Transgenic technology can be used to change the genetic traits of maize, and many trait transformation methods are commonly used [3][4][5][6]. At present, the commonly studied traits include insect pest and herbicide resistance.
In 1996, the United States first approved the commercialization of Cry1Ab GM maize (“Bt176”, “MON810” and “Bt11”), with GM maize now having been promoted and applied for 27 years [7]. As of this publication, 244 GM maize varieties have been approved for cultivation [8]. According to a report by the International Service for the Application of Agricultural Biotechnology (ISAAA), GM crops are grown in 29 countries as of 2019. The planting area of GM maize was 6.09 × 107 hm2 in 2019, accounting for 32% of the total planting area of GM crops. The planting area was the largest in the United States at 3.317 × 107 hm2, followed by Brazil and Argentina [9]. The main use of GM maize globally is used for animal feed [10][11] or as an industrial raw material to extract alcohol. Only a small proportion is consumed directly by humans. The GM maize used for food is mainly used to extract maize oil [12], to make maize syrup, maize flour, or other maize ingredients [13], especially maize starch, which is widely used as a thickener, gelling agent, filler, and water-retention agent in the food industry [14]. GM maize used for food is also directly used as a raw material for food production and processing, such as in the production of tortillas in Mexico [15]; white maize in South Africa, where it is a staple food for most people [16], and maize flakes, popcorn, and maize-related snacks [17]. There is no doubt that GM maize is integrated into the lives of humans worldwide and it is, therefore, important to effectively regulate its production and processing.
The European Union (EU) has adopted a traceability management system and a mandatory labeling system for GM products in the market. The EU Regulation 1830/ 2003/EC, which came into effect in April 2004, stipulates that products containing more than 0.9% (mass fraction, % m/m) must be labeled with the words “genetic improvement” or “processed from a GM crop”. Moreover, the ratio of the DNA copy number should be used as the expression method. The measurement result of the standard substance, based on the mass percentage, shall prevail, and the measurement result of the copy number percentage (DNA copy number ratio, % cpT/cpE) shall be converted into a mass percentage [18]. In China [19], North America [20][21], Australia, New Zealand [22], India [23], and other countries, there are laws and regulations to clarify the labeling management system for GM products. This ensures that business operators and consumers have access to accurate information, so that they can effectively exercise their freedom of choice and be able to control and verify label claims. The latter requirement makes it a necessity to detect the presence of GM organisms (GMOs) through reliable detection methods. The detection of GM crops can be roughly divided into the detection of nucleic acids, proteins, and metabolites, according to the target. In biological cells, DNA is relatively stable compared to proteins, and is not easily destroyed, even after the crops are processed, such that trace or detectable amounts of DNA fragments may remain in the product. Therefore, DNA detection is often the preferred method for identifying GM components of crops. In common polynucleotide detection methods, targets such as promoters (e.g., Cauliflower mosaic virus (CaMV) 35S promoter [24]), terminators (e.g., the nopaline synthase terminator (T-nos) [25][26]), or marker genes (CP4-EPSPS and pat [27][28]) are usually invoked as surrogate markers for transformation. However, methods for testing the above-mentioned target genes cannot distinguish between different strains of GM crops with the same exogenous gene transferred, and the specificity is low. Therefore, a pair of primers spanning the junction of the inserted transgene and the flanking DNA is often used to identify transgene-specific events [29][30], that is, the strain of the GM crop, by detecting the connecting region of the foreign gene and the plant genome. At present, this method is widely used to identify GM maize lines, such as MON810, NK603 [31], Bt11, TC1507, GA21 [32], and so on.
Generally, nucleic acid detection needs to go through a process of nucleic acid extraction and purification, and target detection to obtain results. The purity and quality of the extracted nucleic acid determine the effectiveness of the subsequent amplification, and the setting of the target is the basis for the specificity of detecting the GM maize sample. The method of obtaining the result is also based on intuition and convenience, according to the needs of different detection scenarios. For example, simple and rapid on-site testing methods are crucial for regulating the import and export of GM crops. In this context, the manner in which representative samples are collected, and the timing and reliability of the analysis, is critical for the smooth implementation of regulations and market surveillance.
2. Nucleic Acid Extraction Technology
DNA-based assays are used to examine the transgenic status of processed products, but the quality of the DNA must be ensured. In the study of molecular biology detection methods of different kinds of materials, the choice of DNA extraction methods directly affects its performance and utility. At present, the common extraction methods of plant genomic DNA are sodium dodecyl sulfate (SDS) [33], cetyl trimethylammonium bromide (CTAB) [34], urea extraction method, Chelex-100 method [35], alkali lysis method, polyvinyl pyrrolidone-40 (PVP-40) [36], high-salt and low-pH extraction method [37], and commercial kits [38], etc. These DNA extraction methods are generally consistent in principle, involving cell lysis for DNA release and the removal of impurities for DNA purification (Figure 1). However, the content of proteins, polysaccharides, phenols, and other substances in different plant materials is not the same, which causes great difficulties in the extraction, separation, and purification of DNA. For plant-derived samples, the appropriate method should be selected according to the characteristics of the sample, and the methods may need to be adjusted and optimized.
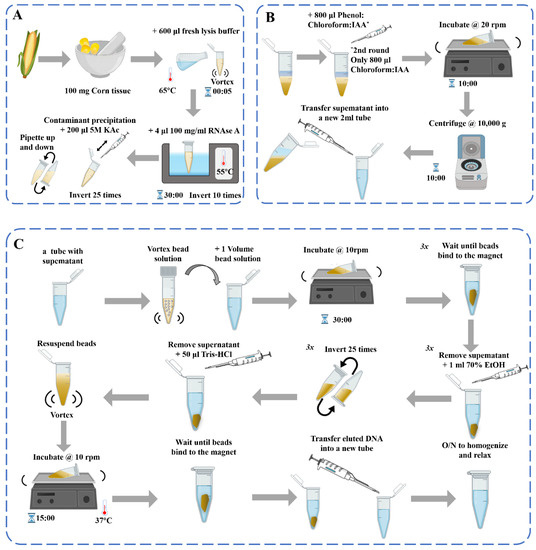
Figure 1. Schematic diagram of the DNA extraction process [39]: (A) DNA extraction; (B) Contaminant precipitation and Phenol: Chloroform purification 2×; (C) Genomic DNA purification with beads.
In the process of DNA extraction of maize samples, there are related extraction methods of maize raw materials for various plant tissues, such as pollen, leaves, maize silk [40], endosperm, and others [41]. Maize is an angiosperm, and its fruit has a unique structure, mainly composed of a pericarp, seed coat, endosperm, and embryo (germ, radicle, hypocotyl, and cotyledon). The pericarp and seed coat of sexually reproduced maize are developed from the ovary wall and the integument of the female parent, and all of the genetic material comes from the female parent. The pericarp and seed coat of mature maize almost grow together and are difficult to separate. The endosperm develops from the fertilized polar nucleus, and its genetic material is different from that of the embryo and adult plant; therefore, proper consideration should be given when extracting raw materials. The traditional gene-screening method involves extracting genomic DNA from the leaves [42][43] (or young leaves) of individual plants after planting large populations of maize in the field. The single-seed maize DNA extraction method mainly involves crushing the seeds or using the seed endosperm to extract DNA. DNA extraction methods for crude maize products are mainly used in the detection of GMOs in feed [44][45]. According to the degree of DNA degradation from smallest to greatest, the relevant foods can be divided into raw maize, frozen maize, canned maize, and dry packet maize samples [46]. The deep-processed corn products are mainly seasoning, puffed, fried, saccharified, and fermented flour products, such as maize oil [47], maize starch, maize chip, popcorn, maize stick, crisp maize horn, wowtou [48], etc.
Cell lysis, including mechanical, enzymatic, and chemical lysis, is an essential process for DNA extraction from cell tissues. Mechanical cracking, such as grinding in liquid nitrogen, heat shock, homogenization, and ultrasonic treatment [49], may lead to DNA breakage, which generally plays an auxiliary role in practical applications. The ultrasonic intensity and gap time need to be strictly controlled to avoid excessive DNA breakage. Enzymatic cleavage uses specific enzymes (such as pectinases) to destroy cells and release nucleic acids that can be extracted in conjunction with chemical cleavage.
CTAB and SDS are the two most commonly used and effective chemical lysing agents. In general, surfactants such as CTAB and SDS tend to interact with polymers (e.g., proteins and DNA) driven by electrostatic, diaxial, and hydrophobic forces [50]. CTAB can dissolve cell membranes, and in high-salt (>0.7 mol/L NaCl) solutions, CTAB can form soluble and stable complexes with proteins and polysaccharides, but cannot precipitate nucleic acids [51]. In low-salt (0.1~0.5 mol/L NaCl) solutions [52], SDS can, under alkaline conditions of 55~65 °C, lyse cells and make DNA free, while denaturing proteins and binding to them [53]. Recently, ionic liquids (ILs) and magnetic ionic liquids (MIL) have been explored as novel solvents for extracting DNA from complex biological matrices [54]. ILs and MILs promote DNA extraction through electrostatic interactions between cationic and negatively charged phosphate backbones, and hydrophobic interactions between the alkyl chains of the solvent and DNA bases [55][56]. Microscale electroporation is an emerging technology for the release of intracellular materials [57]. Its mechanism of action is that, when the electric field intensity of the applied electric pulse reaches a certain order of magnitude, the cell membrane undergoes a configurational change, and a large number of micropores appear. This increases the permeability of the cell membrane, which is conducive to the release of various macromolecular substances (such as DNA, RNA, proteins, chemical small molecules, etc.). However, adjusting the applied voltage and pulse length to determine the optimal conditions for cell lysis and DNA extraction still requires experimental verification.
Crude DNA extracts contain large amounts of protein, RNA, sugars, and other impurities; therefore, DNA purification is essential. Most proteins can be removed by denaturation and precipitation after treatment with chloroform or phenol, which are common methods of protein removal [58]. The alternating use of phenol and chloroform, two different protein denaturants, can enhance the effect of protein removal [49]. It should be noted that these chemicals, phenol and chloroform, have certain oxidizing properties and can seriously damage DNA if used improperly. For example, guanine is particularly sensitive to oxidation, and exposure to phenol/chloroform can result in the formation of 8-oxoguanine [59]. Furthermore, phenol and chloroform are volatile and toxic, with chloroform classified as “reasonably expected to be a human carcinogen based on sufficient evidence of carcinogenicity in laboratory animal studies”, according to a U.S. Department of Health and Human Services report on carcinogens [60].
RNA can be removed by digestion with RNase A for 1~2 h at approximately 37 °C, or DNA can be purified by cesium chloride density gradient centrifugation, which results in a high-quality DNA preparation [58]. Maize plant cells have thickened secondary walls and large vacuoles that store a large number of secondary substances, such as polysaccharides and polyphenols. The surfactant CTAB is better than SDS at removing polysaccharides [61][62]. Appropriately increasing the content of CTAB (according to the actual situation, such as increasing it to 3%, but not using less than 1% [63]) β-mercaptoethanol (0.2%–1%, determined according to the actual situation) [64] can effectively remove polysaccharides and other secondary biomolecules. Simultaneously, the polyphenols contained in plants are oxidized under the catalytic action of polyphenol oxidase, resulting in a lower quality of the extracted DNA. The main methods to remove the effects of polyphenols include adding antioxidants to the extraction medium or adding PVP or ascorbic acid during grinding [65]. Further oxidation of phenolic compounds can be prevented by using a higher concentration of salt and a less-acidic medium [66]. Commercial solid-phase extraction kits with silica-based centrifugal columns have been developed to standardize procedures and make them more efficient. These kits use cleavage buffers containing CTAB or SDS, binding buffers consisting of dissociative salts to facilitate DNA adsorption onto silica adsorbents, and washing buffers containing organic solvents to eluate and purify the DNA [67]. The entire extraction process is conducted at room temperature, the rigor of the experimental requirements is low, and the concentration and purity of the DNA obtained meet the requirements of most contemporary molecular biology applications. Currently, commercial kits used for plant DNA extraction also employ functionalized magnetic materials to simplify the purification step through the use of external magnets.
For DNA extraction of GM maize, the traditional CTAB method is the most suitable method for extracting amplifiable DNA from highly processed maize gluten, which is often used as a protein-rich feed ingredient. This method can produce sufficient amounts of amplified DNA in laboratory tests to control the compliance of the tested substrate with event tolerance limits and labeling thresholds for authorized GM maize [44]. The SDS extraction method often results in a higher DNA yield, better cell lysis efficiency, a lower DNA shear rate, and higher diversity than the CTAB method [68]. Therefore, among the many DNA extraction methods at present, new improvements of both the CTAB and SDS methods are currently under development [53][69].
With rapid developments in the field of molecular biology, precision diagnosis, and treatment, the demand for emerging nucleic acid extraction technologies with high throughput, purity, and quality is constantly increasing. As a simple, fast, reliable and automated nucleic acid extraction method, the magnetic beads(MBs) method [70] for nucleic acid extraction has attracted more and more attention. The process of DNA extraction by the MBs method is simple, without repeated centrifugation, column separation, or vacuum filtration [71]. The entire process consists of four steps including lysis, binding, separation, and elution. Therefore, the process is fast and the extraction efficiency is high, culminating in extracted nucleic acids that are high in purity and concentration. Moreover, it is safe and non-toxic, does not use toxic reagents (such as phenol, etc.), reduces personnel hazards, and can be easily adapted for automated batch operation [72]. MBs and automatic nucleic acid extraction instruments can be used for high-throughput automated extraction of nucleic acids from large numbers of clinical samples. MBs with functionalized surfaces that can capture nucleic acids have been widely used to extract nucleic acids from biological samples, and multiple forms have been developed [73]. Examples include manual extraction using magnetic frames or microfluidic chips [74], automated robotic processing, and also the separation of ctDNA by superparamagnetic bead particles in microfluidic platforms for early cancer detection, etc. Combining them with traditional gene extraction methods can efficiently and quickly extract plant nucleic acids, and has an absolute advantage over other methods in the detection of low-content genetically modified components. Sebastian et al. [75] used centrifugal microfluidic technology, using continuous rotating magnetophoresis to facilitate magnetic bead integration and nucleic acid extraction where needed. This solution solves the drawbacks of magnetic bead-based solid-phase extraction that may cause nucleic acid loss due to the handling of magnetic beads when they are transferred from one chamber to another, resulting in higher yield and purity in the end. Jiang et al. [70] and others established a no-elution MB-based nucleic acid extraction method by introducing PEPPG F68 into the lysate and using NaOH solution instead of alcohol as the washing buffer. It avoids the dilution and loss of the target nucleic acid during the elution process, as well as the possible loss of sensitivity and false-negative results. At the same time, the detection sensitivity of loop-mediated isothermal amplification (LAMP) is significantly improved, which has broad application prospects. The current demand by molecular biologists is for convenient, rapid, and inexpensive DNA extraction and detection methods and more compact, portable equipment options to enhance real-time capabilities. The merging of extraction and microflow body technologies [76] to automate nucleic acid detection [77][78] is also in high demand.
3. Traditional Detection Technology
3.1. Variable-Temperature Amplification
Event-specific polymerase chain reaction (PCR) targeting unique sequences spanning the insert DNA and flanking genomic DNA has the highest level of specificity and is commonly used to confirm the identity and authorization status of GMO ingredients and to quantify GMO content [79]. DNA-based PCR methods are considered the most reliable and versatile techniques for the identification and quantification of GMOs, with the chief method being variable-temperature amplification. Variable-temperature amplification technology includes three steps, high-temperature denaturation, low-temperature annealing, and a suitable-temperature extension, and generally refers to PCR technology and its derivatives, such as real-time fluorescent PCR [80][81], droplet digital PCR (ddPCR) [82], nested or semi-nested PCR [83], multiplex PCR [84], etc.
The PCR method is the most commonly used molecular detection technique and is the standard method for detecting GMOs [85]. Standard qualitative PCR is the most commonly used PCR detection method. The mechanism is as follows: after the primer and template DNA are specifically combined in accordance with the principle of complementary base pairing, under the catalysis of Taq DNA polymerase, using deoxyribonucleotide triphosphates (dNTPs) as the raw material, a new DNA strand is synthesized according to the principle of semi-conservative replication. After “n” times of amplification, the total number of progeny DNA is 2n, and finally achieves a million-fold amplification of the number of target DNA fragments, which facilitates the detection of subsequent target DNA fragments [86] and, finally, achieves a million-fold amplification of the number of target DNA fragments, which facilitates the detection of subsequent target DNA fragments. Standard PCR is used for the amplification of transgenic crop genes owing to its simple operation, high efficiency, and low cost. Quantitative PCR (qPCR), based on a standard curve, is considered the gold standard technique for the analysis of GMOs because of its high sensitivity and good stability [87]. Real-time fluorescent qPCR [88] is a technology that adds fluorescent groups to the PCR reaction system and uses the accumulation of fluorescent signals to monitor the entire PCR reaction process in real time. This technology uses the strength of the fluorescent signal to determine the number of specific amplification products over time, and an unknown template is quantitatively analyzed using a standard curve. This technology can quantitatively analyze DNA templates and has the characteristics of high sensitivity, specificity, and reliability; low pollution; and timely and accurate detection. It can perform both absolute and relative quantifications and is widely used to inspect GM maize [89]. Various forms of qPCR are constantly being developed to meet the needs of practical applications, focusing on duplex and multiplex reactions to improve detection throughput and efficiency.
The concept of digital PCR (dPCR) was first proposed by Vogelstein et al. in 1999 [90]. In dPCR, the reaction mixture is divided into many individual reactions called partitions, and each reaction does not contain one or more copies of the target. Reads are partitioned as negative or positive at the endpoints, and DNA concentrations are calculated using a Poisson distribution [91]. The partitioning of reaction volumes using wells on a chip in microfluidics/chip-based dPCR [92] and droplets in emulsion/ddPCR [93] are the two main approaches. In cdPCR, reactions are divided into hundreds or thousands of chambers in a single plate or array. Many studies have used chip-based platforms, such as the microwell chip-based QuantStudio 12k flex dPCR and 3D dPCR (Life Technologies), for the detection of GMOs in the field [94][95]. The Constellation system (Formulatrix) is a plate-based microfluidic dPCR system that offers five-color multiplexing. The biggest difference between the cdPCR platforms is the number of partitions created per sample and the number of samples analyzed in one run [96]. ddPCR has a synergistic effect on droplet microfluidics. It improves the sensitivity of PCR at the single-molecule level by dividing tens of microliters of PCR mixture into tens of thousands of droplets and it can perform absolute quantification without a standard curve, thus avoiding the amplification efficiency bias observed in qPCR. It enables accurate target determination even at low copy numbers and can be significantly cost-effective when combined with multiplexing [97]. However, the droplet reaction generator used for ddPCR is bulky and complicated, which is an important limitation for its use in on-site detection. Thus far, microfluidic platforms for droplet generation using centrifugal forces, such as those utilizing ferrofluids, electromagnets [97], and surface acoustic waves [98]. Using the working principle of the indirect pressurization method, Park et al. [99] developed a pushbutton-activated microfluidic dropenser (droplet dispenser). Its use for sample preparation in ddPCR eliminates the need for benchtop droplet generators and automated pipetting, making ddPCR an on-site molecular diagnostic tool. In conclusion, dPCR has proven to be an effective tool for the quantification of maize and soybean GMOs and GMOs in complex matrix samples with precision and accuracy the same as or better than qPCR methods [100]. Recently developed multiplex dPCR methods [87][101] may be useful for analyzing samples containing multiple genetic modification events.
More types of PCR technologies are constantly being developed to meet the requirements for on-site rapid detection and high throughput. The use of an ultrafast PCR system can significantly reduce PCR run times and the number of reagents required for analysis. Therefore, ultrafast PCR systems have recently been studied and applied in various fields. The latest example of an ultrafast PCR system is a system used in rice detection [102]. The analysis principle is the same as that of real-time fluorescent qPCR, based on SYBR green [103], except that Evagreen dye is used as the intercalating dye instead of SYBR green. It requires 18% of the detection time of traditional PCR and 23% of the detection time of real-time PCR, and it can support small portable analyzers, thus providing a new strategy for the on-site detection of GM maize.
3.2. Isothermal Amplification
Isothermal nucleic acid amplification technology is used for nucleic acid amplification at a constant temperature. According to the different methods of single-stranded template formation, isothermal amplification can be divided into the following four categories: (1) strand-displacing DNA polymerase-mediated reactions such as Loop-Mediated Isothermal Amplification (LAMP) [104], Rolling Circle Amplification (RCA) [105], Cross-Primed Amplification (CPA) [106], Nucleic Acid Sequence-Based Amplification (NASBA [107]), and Multiple Displacement Amplification (MDA) [108]; (2) Enzymatic unwinding primer annealing reaction, such as Helicase-Dependent Amplification (HDA) [109], Recombinase Polymerase Amplification (RPA) [110], and Ligase Chain Reaction (LCR) [111]; (3) RNA transcription-based amplification, such as Transcription Mediated Amplification (TMA); and (4) Requires reactions assisted by single-strand cleavage enzymes, such as Strand Displacement Amplification (SDA) [112], and Isothermal Strand Displacement Amplification (iSDA) [113]. LAMP, RPA, and CPA are widely used for the detection of GM crops.
LAMP employs a DNA polymerase and a set of four specially designed primers that recognize a total of six different sequences in the target DNA. Internal primers containing the sequences of the sense and antisense strands of the target DNA initiate LAMP [104]. The product is a mixture of stem-loop DNA with stems of various sizes and cauliflower-like structures. Multiple loops are induced by annealing between alternating inverted repeats of the target sequence in the same strand. This enables simpler and more selective detection. For example, through a mechanism similar to multivalent antigen antibody interactions, the target sequence has a higher degree of specificity [104]. This method is insensitive to inhibitors, and can be used with crude DNA samples. LAMP is a simple and reliable GM detection method that can be performed on a thermal cycler, heating block, or portable constant temperature real-time amplification system. After the reaction is completed, using nucleic acid staining or fluorescent dyes, such as SYBR® Green and hydroxynaphthol blue, the LAMP products can be visualized and monitored using turbidity analysis or real-time LAMP [114]. Since the first report on the LAMP method in 2000, the number of studies using LAMP to detect GM ingredients has increased annually [115][116][117].
RPA technology was first proposed in 2006 by Piepenburg et al. [118]. In RPA, isothermal amplification of specific DNA fragments is achieved by the binding of reverse oligonucleotide primers to the template DNA and extension via DNA polymerase. It does not require the orientation of the primers to their complementary target sequences. RPA can be used to amplify cDNA generated by the reverse transcription of double-stranded DNA, single-stranded DNA, methylated DNA, RNA, or miRNA, and multiple reverse transcriptases are used for RPA [110]. When using RPA directly in milk [119] or seed powder [120], only thermal lysis, nuclease-free water lysis, or EzWayTM Direct PCR buffer are required to release the desired nucleic acid. With the assistance of a variety of enzymes, the in vitro amplification of nucleic acid can be completed at a constant temperature of 31–37 °C for 20 minutes [110]. Traditional in vitro nucleic acid amplification techniques do not have a rapid response, high sensitivity, high specificity, or low equipment dependence. Compared with SDA, RCA, and LAMP, RPA does not require an initial denaturation step to generate single-stranded (ss)DNA from double-stranded (ds)DNA targets, highlighting its suitability for use in the field [121]. In 2014, a commercial RPA kit launched by the British company, Twist DX [122], made the detection more convenient. Simultaneously, a variety of probes can be combined to expand the application range of RPA technology. RPA appears to be particularly well suited for multiplexing, where different targets can be verified with different efficiencies; however, it currently requires laborious optimization steps. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPER-associated protein [123], lateral flow assays [124][125] and microfluidics [126] have provided additional options for the on-site point-of-care detection of transgenes (Figure 2).
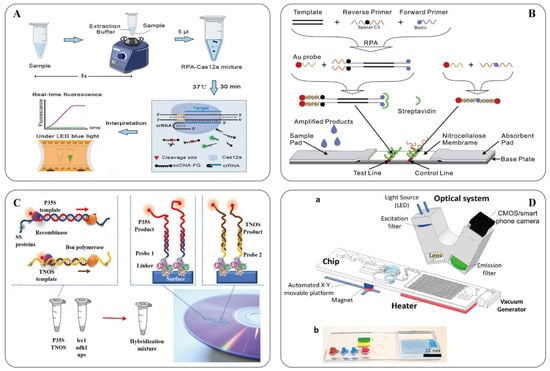
Figure 2. (A) Schematic showing the ORCD assay system for rapid and visual nucleic acid detection [123]; (B) Schematic of the LFNAA (lateral flow nucleic acid assay) [125]; (C) Scheme of the assay for GMOs detection based on multiplex RPA amplification (left) and hybridization assay in the array format (right) [127]; (D) Microfluidic platform for sample-to-answer digital RT-RPA [126]: (a) Schematic illustration of the total system consisting of a magnetic platform, heater unit, vacuum generator, detection part with camera, and light-emitting diode (LED). The disposable chip has three operational zones to prepare and mix the reagents and detect the pathogen agent. (b) Dye-loaded chambers for visualization of chips. Brown, blue, and red show lysis, washing and elution chambers, respectively, for sample preparation. Green and yellow chambers show RPA mixture and mineral oil chambers, respectively.
CPA technology is an isothermal DNA amplification system developed by Us-Tar Biotechnology Co., Ltd [106]. The system relies on only one ring structure for replication [128]. The CPA assay enables the amplification of nucleic acid sequences at a constant temperature and requires only an enzyme with strand displacement activity and a set of five primers to perform the CPA reaction, without the need for an initial denaturation step or the addition of a nickase [129]. At an assay temperature of 63 °C, the formation of primer template hybrids under transient spontaneous denaturing bubbles in the DNA template are more favorable than the re-annealing of the template strands by high concentrations of primers relative to the template DNA. Strand displacement is facilitated by annealing cross-primers with 5’ ends that are not complementary to the template strand and the binding of displacement primers upstream of the cross-primers. The resulting exponential amplification of the target DNA is highly specific and sensitive [106]. CPA has traditionally yielded results through expensive fluorescence-based techniques, tedious gel electrophoresis procedures, or the measurement of turbidity using a spectrophotometer. These methods require complex and bulky optics or exposure to carcinogenic dyes, which limits their wider use in resource-limited laboratories [128]. In addition, colorimetric indicators such as pH-sensitive dyes (neutral red), malachite green (MG), and hydroxynaphthol blue (HNB) [130] were also used as complementary techniques to monitor CPA responses.
3.3. Gene Chip Technology
Gene chip technology is a GM food detection technology developed by the American company Affymetrix in the 1990s. It has rapidly developed as a high-tech molecular biology tool in recent years. Gene chips are also known as DNA chips or microarrays [131]. This technology involves arranging a large number of DNA fragments or oligonucleotide fragments at an orderly density on a solid-phase carrier, using specific probes on the surface of the solid-phase carrier to hybridize with labeled samples, and using a chip scanner to detect and analyze hybridization signals [132]. It can accurately detect different types of DNA sequences in samples qualitatively and quantitatively. When microarray hybridization is used for gene analysis, information on differences in gene expression can be obtained from very few experimental samples. When using this technology for detection, the sample is pretreated and purified to obtain a highly pure DNA sample, which is then amplified by PCR, labeled with fluorescence, and hybridized with the DNA probe on the gene chip. The signal is then read to obtain the result [133]. Gene chips can immobilize a large number of oligonucleotide probes for different target genes; therefore, gene chip technology can detect dozens or even hundreds of genes simultaneously [134] and can detect multiple components in GM foods. Lu et al. [135] used a gene chip detection method combined with multiplex PCR to simultaneously detect multiple pairs of genes on a single chip. Seven types of GM maize components were detected: Bt176, Bt11, GA21, Mon810, Mon863, TC1507, and NK603. This method greatly improves the accuracy and efficiency of detection, with a sensitivity as high as 0.01%. Turkec et al. [131] tested 1830 different probes and developed a high-density oligonucleotide microarray platform for 12 GM varieties (nine maize and three soybean varieties). This method eliminates the need for a PCR amplification step, which simplifies the analysis and allows the quantification of each detected GMO, enabling the specific detection of each GM crop with a sensitivity of 1% (DNA concentration).
Gene chip technology offers advantages, such as a high level of parallelism, high throughput, high specificity, high sensitivity, and automation. However, due to the late start of the development of this technology, strong comprehensiveness, strong professionalism, and high cost, the process of making gene chips is relatively complicated, and there are certain problems, such as background interference. Therefore, the popularization and promotion of this technology in practical applications are limited [136]. At present, the types of GM edible agricultural products grown worldwide is limited, and the detection of GM ingredients often only requires the detection of dozens of target genes, whereas gene chip technology can perform multigene or even whole-gene detection [134]. Therefore, considering the experimental cost and utilization rate, this technology is currently unsuitable for transgene detection. In general, gene chip technology requires extremely small samples and has the advantages of being fast, time-saving, pollution-free, accurate, and suitable for automated operation; however, current research using this technology is in the field of disease diagnosis and microbial detection, and research on transgenic detection is focused more on microfluidic chips [137]. Microfluidic chips integrate multiple operating platforms into one chip through micro-processing technology, with less consumption of samples and reagents, a fast reaction speed, and a large number of parallel processes. Therefore, it has greater development potential.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241512247
References
- FAOSTAT. Available online: https://www.fao.org/faostat/es/#data/QCL (accessed on 21 May 2023).
- Spatial-Temporal Evolution of Scientific Production about Genetically Modified Maize. Agriculture 2021, 11, 246.
- EFSA Panel on Genetically Modified Organisms (GMO); Naegeli, H.; Birch, A.N.; Casacuberta, J.; De Schrijver, A.; Gralak, M.A.; Guerche, P.; Jones, H.; Manachini, B.; Messéan, A.; et al. Scientific Opinion on an Application by DOW AgroSciences LLC (EFSA-GMO-NL-2010-89) for Placing on the Market the Genetically Modified Herbicidetolerant Maize DAS-40278-9 for Food and Feed Uses, Import and Processing under Regulation (EC) No 1829/2003. EFSA J. 2016, 14, e04633.
- Sutradhar, M.; Mandal, N. Reasons and Riddance of Agrobacteriumtumefaciens Overgrowth in Plant Transformation. Transgenic Res. 2023, 32, 33–52.
- Dong, O.X.; Ronald, P.C. Targeted DNA Insertion in Plants. Proc. Natl. Acad. Sci. USA 2021, 118, e2004834117.
- Muppala, S.; Gudlavalleti, P.K.; Malireddy, K.R.; Puligundla, S.K.; Dasari, P. Development of Stable Transgenic Maize Plants Tolerant for Drought by Manipulating ABA Signaling through Agrobacterium-Mediated Transformation. J. Genet. Eng. Biotechnol. 2021, 19, 96.
- Snow, A.A.; Palma, P.M. Commercialization of Transgenic Plants: Potential Ecological Risks. BioScience 1997, 47, 86–96.
- Maize (Zea mays L.) GM Events|GM Approval Database—ISAAA.Org. Available online: https://www.isaaa.org/gmapprovaldatabase/crop/default.asp?CropID=6&Crop=Maize (accessed on 13 July 2023).
- Global Status of Commercialized Biotech/GM Crops: 2019—ISAAA Brief 55-2019|ISAAA.Org. Available online: https://www.isaaa.org/resources/publications/briefs/55/default.asp (accessed on 13 July 2023).
- Anderson, J.A.; Herman, R.A.; Carlson, A.; Mathesius, C.; Maxwell, C.; Mirsky, H.; Roper, J.; Smith, B.; Walker, C.; Wu, J. Hypothesis-Based Food, Feed, and Environmental Safety Assessment of GM Crops: A Case Study Using Maize Event DP-202216-6. GM Crops Food-Biotechnol. Agric. Food Chain 2021, 12, 282–291.
- Avsar, B.; Sadeghi, S.; Turkec, A.; Lucas, S.J. Identification and Quantitation of Genetically Modified (GM) Ingredients in Maize, Rice, Soybean and Wheat-Containing Retail Foods and Feeds in Turkey. J. Food Sci. Technol. 2020, 57, 787–793.
- Hashemzadeh, H.; Karbasi, A.; Mohammadi, H.; Firoozzare, A.; Boccia, F. Investigating the Effect of Nudges on Consumers’ Willingness to Pay for Genetically Modified Corn Oil. Sustainability 2022, 14, 12705.
- Boccia, F.; Punzo, G. A Choice Experiment on Consumer Perceptions of Three Generations of Genetically Modified Foods. Appetite 2021, 161, 105158.
- Zhang, R.; Ma, S.; Li, L.; Zhang, M.; Tian, S.; Wang, D.; Liu, K.; Liu, H.; Zhu, W.; Wang, X. Comprehensive Utilization of Corn Starch Processing By-Products: A Review. Grain Oil Sci. Technol. 2021, 4, 89–107.
- Delgado-Valerio, P.; Ramón-Amado, A.; Piñeyro-Nelson, A.; Álvarez-Buylla, E.R.; Ayala-Angulo, N.M.; Molina-Sánchez, A. Presencia de secuencias transgénicas en masa para tortillas de poblados urbanos y rurales de la meseta purépecha, michoacán, méxico. Rev. Fitotec. Mex. 2022, 45, 283.
- Ala-Kokko, K.; Nalley, L.L.; Shew, A.M.; Tack, J.B.; Chaminuka, P.; Matlock, M.D.; D’Haese, M. Economic and Ecosystem Impacts of GM Maize in South Africa. Glob. Food Secur. Agric. Policy Econ. Environ. 2021, 29, 100544.
- Brara, Z.; Costa, J.; Villa, C.; Grazina, L.; Bitam, A.; Mafra, I. Surveying Genetically Modified Maize in Foods Marketed in Algeria. Food Control 2020, 109, 106928.
- EUR-Lex. Regulation (EC) No 1830/2003 of the European Parliament and of the Council of 22 September 2003 Concerning the Traceability and Labelling of Genetically Modified Organisms and the Traceability of Food and Feed Products Produced from Genetically Modified Organisms and Amending Directive 2001/18/EC; Publications Office of the EU: Luxembourg, 2003; Volume 268.
- Decree of the Ministry of Agriculture of the People’s Republic of China (No. 10) Measures for the Administration of the Labeling of Agricultural Genetically Modified Organisms__State Council Gazette No. 2002 of 35_Chinese Government Website. Available online: https://www.gov.cn/gongbao/content/2002/content_61835.htm (accessed on 13 July 2023).
- 7 CFR Part 66—Part 66—National Bioengineered Food Disclosure Standard. Available online: https://www.law.cornell.edu/cfr/text/7/part-66 (accessed on 13 July 2023).
- Branch, L.S. Consolidated Federal Laws of Canada, Food and Drug Regulations. Available online: https://laws-lois.justice.gc.ca/eng/regulations/C.R.C.,_c._870/page-54.html#h-574622 (accessed on 13 July 2023).
- Federal Register of Legislation—Australian Government. Available online: https://www.legislation.gov.au/Series/F2015L00404 (accessed on 13 July 2023).
- FSSAI. Available online: https://fssai.gov.in/cms/food-safety-and-standards-regulations.php (accessed on 13 July 2023).
- Genesiska; Suratmi, R.C. Detection of Promoter Designed for Transgenic Plant in Local Soybean. IOP Conf. Ser. Earth Environ. Sci. 2020, 458, 012011.
- He, Y.; Fan, Z. A Novel Biosensor Based on DNA Hybridization for Ultrasensitive Detection of NOS Terminator Gene Sequences. Sens. Actuators B Chem. 2018, 257, 538–544.
- Zadeh, R.B.; Safaeian, S.; Moslemi, E.; Nadushen, R.M.; Esfahani, K. Monitoring of Infant Formula and Baby Food for the Pat and NOS Terminator of Genetically Modified Maize and Soybean by Real-Time PCR in Iran. Iran. J. Pharm. Res. 2022, 21, e126921.
- Jiao, P.; Jin, S.; Chen, N.; Wang, C.; Liu, S.; Qu, J.; Guan, S.; Ma, Y. Improvement of Cold Tolerance in Maize (Zea mays L.) Using Agrobacterium-Mediated Transformation of ZmSAMDC Gene. GM Crops Food-Biotechnol. Agric. Food Chain 2022, 13, 131–141.
- Zeng, H.; Wang, J.; Jia, J.; Wu, G.; Yang, Q.; Liu, X.; Tang, X. Development of a Lateral Flow Test Strip for Simultaneous Detection of BT-Cry1Ab, BT-Cry1Ac and CP4 EPSPS Proteins in Genetically Modified Crops. Food Chem. 2021, 335, 127627.
- Xu, J.-M.; Zhu, J.-S.; Li, M.-Z.; Hu, H.; Mao, C.-Z. Progress on Methods for Acquiring Flanking Genomic Sequence. Yi Chuan Hered. 2022, 44, 313–321.
- Siddique, K.; Wei, J.; Li, R.; Zhang, D.; Shi, J. Identification of T-DNA Insertion Site and Flanking Sequence of a Genetically Modified Maize Event IE09S034 Using Next-Generation Sequencing Technology. Mol. Biotechnol. 2019, 61, 694–702.
- Li, X.; Shen, K.; Yang, L.; Wang, S.; Pan, L.; Zhang, D. Applicability of a Novel Reference Molecule Suitable for Event-Specific Detections of Maize NK603 Based on Both 5′ and 3′ Flanking Sequences. Food Control 2010, 21, 927–934.
- Bhoge, R.K.; Chhabra, R.; Randhawa, G.; Sathiyabama, M.; Singh, M. Event-Specific Analytical Methods for Six Genetically Modified Maize Events Using Visual and Real-Time Loop-Mediated Isothermal Amplification. Food Control 2015, 55, 18–30.
- Tai, T.H.; Tanksley, S.D. A Rapid and Inexpensive Method for Isolation of Total DNA from Dehydrated Plant Tissue. Plant Mol. Biol. Report. 1990, 8, 297–303.
- Murray, M.G.; Thompson, W.F. Rapid Isolation of High Molecular Weight Plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326.
- Giraffa, G.; Rossetti, L.; Neviani, E. An Evaluation of Chelex-Based DNA Purification Protocols for the Typing of Lactic Acid Bacteria. J. Microbiol. Methods 2000, 42, 175–184.
- Kim, C.S.; Lee, C.H.; Shin, J.S.; Chung, Y.S.; Hyung, N.I. A Simple and Rapid Method for Isolation of High Quality Genomic DNA from Fruit Trees and Conifers Using PVP. Nucleic Acids Res. 1997, 25, 1085–1086.
- Padmalatha, K.; Prasad, M. Optimization of DNA Isolation and PCR Protocol for RAPD Analysis of Selected Medicinal and Aromatic Plants of Conservation Concern from Peninsular India. Afr. J. Biotechnol. 2006, 5, 230–234.
- Bashalkhanov, S.; Rajora, O.P. Protocol: A High-Throughput DNA Extraction System Suitable for Conifers. Plant Methods 2008, 4, 20.
- Russo, A.; Mayjonade, B.; Frei, D.; Potente, G.; Kellenberger, R.T.; Frachon, L.; Copetti, D.; Studer, B.; Frey, J.E.; Grossniklaus, U.; et al. Low-Input High-Molecular-Weight DNA Extraction for Long-Read Sequencing From Plants of Diverse Families. Front. Plant Sci. 2022, 13, 883897.
- Nadeem, S.A.; Mughal, D.; Butt, N.A.; Ahmed, S.; Khan, I.A. Utilization of Corn Silk for GMO Detection Through Real-Time PCR. Waste Biomass Valorization 2023.
- SanJuan-Badillo, A.; Galvez, A.; Plasencia, J.; Quirasco, M. Assessment of DNA extraction methods from various maize (Zea mays L.) tissues for environmental GMO monitoring in Mexico. Part I: Detection by end-point PCR. Agrociencia 2014, 48, 17–33.
- Gao, S.; Martinez, C.; Skinner, D.J.; Krivanek, A.F.; Crouch, J.H.; Xu, Y. Development of a Seed DNA-Based Genotyping System for Marker-Assisted Selection in Maize. Mol. Breed. 2008, 22, 477–494.
- Leach, K.A.; McSteen, P.C.; Braun, D.M. Genomic DNA Isolation from Maize (Zea mays) Leaves Using a Simple, High-Throughput Protocol. Curr. Protoc. Plant Biol. 2016, 1, 15–27.
- Matthes, N.; Westphal, K.; Haldemann, C.; Egert, M.; Jokisch, C.; Speck, B. Validation of a Modified CTAB Method for DNA Extraction from Protein-Rich Maize Feedstuffs. J. Consum. Prot. Food Saf. 2020, 15, 331–340.
- Turkec, A.; Kazan, H.; Karacanli, B.; Lucas, S.J. DNA Extraction Techniques Compared for Accurate Detection of Genetically Modified Organisms (GMOs) in Maize Food and Feed Products. J. Food Sci. Technol. 2015, 52, 5164–5171.
- Takabatake, R.; Noritake, H.; Noguchi, A.; Nakamura, K.; Kondo, K.; Akiyama, H.; Teshima, R.; Mano, J.; Kitta, K. Comparison of DNA Extraction Methods for Sweet Corn and Processed Sweet Corns. Food Hyg. Saf. Sci. 2013, 54, 309–315.
- Kishine, M.; Noguchi, A.; Mano, J.; Takabatake, R.; Nakamura, K.; Kondo, K.; Kitta, K. Detection of DNA in Highly Processed Foods. Food Hyg. Saf. Sci. 2018, 59, 151–156.
- Singh, M.; Sodhi, K.K.; Paliwal, A.; Sharma, S.; Randhawa, G. Efficient DNA Extraction Procedures for Processed Food Derivatives-a Critical Step to Ensure Quality for GMO Analysis. Food Anal. Methods 2021, 14, 2249–2261.
- Liu, Y.; Liu, X.; Cui, Y.; Yuan, W. Ultrasound for Microalgal Cell Disruption and Product Extraction: A Review. Ultrason. Sonochem. 2022, 87, 106054.
- Xia, Y.; Chen, F.; Du, Y.; Liu, C.; Bu, G.; Xin, Y.; Liu, B. A Modified SDS-Based DNA Extraction Method from Raw Soybean. Biosci. Rep. 2019, 39, BSR20182271.
- Jones, A.S. The Isolation of Bacterial Nucleic Acids Using Cetyltrimethylammonium Bromide (Cetavlon). Biochim. Biophys. Acta 1953, 10, 607–612.
- Masoodi, K.Z.; Lone, S.M.; Rasool, R.S. Chapter 7—Genomic DNA Extraction from the Plant Leaves Using the CTAB Method. In Advanced Methods in Molecular Biology and Biotechnology; Masoodi, K.Z., Lone, S.M., Rasool, R.S., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 37–44. ISBN 978-0-12-824449-4.
- Chabi Sika, K.; Kefela, T.; Adoukonou-Sagbadja, H.; Ahoton, L.; Saidou, A.; Baba-Moussa, L.; Jno Baptiste, L.; Kotconi, S.O.; Gachomo, E.W. A Simple and Efficient Genomic DNA Extraction Protocol for Large Scale Genetic Analyses of Plant Biological Systems. Plant Gene 2015, 1, 43–45.
- Wang, X.; Liu, M.; Ding, X. Guanidinium Hydrophobic Magnetic Ionic Liquid-Based Dispersive Droplet Extraction for the Selective Extraction of DNA. Langmuir 2021, 37, 11665–11675.
- Emaus, M.N.; Cagliero, C.; Gostel, M.R.; Johnson, G.; Anderson, J.L. Simple and Efficient Isolation of Plant Genomic DNA Using Magnetic Ionic Liquids. Plant Methods 2022, 18, 37.
- Marengo, A.; Cagliero, C.; Sgorbini, B.; Anderson, J.L.; Emaus, M.N.; Bicchi, C.; Bertea, C.M.; Rubiolo, P. Development of an Innovative and Sustainable One-Step Method for Rapid Plant DNA Isolation for Targeted PCR Using Magnetic Ionic Liquids. Plant Methods 2019, 15, 1–11.
- Choi, S.-E.; Khoo, H.; Hur, S.C. Recent Advances in Microscale Electroporation. Chem. Rev. 2022, 122, 11247–11286.
- Wu, X.; Gong, F.; Wang, W. Protein Extraction from Plant Tissues for 2DE and Its Application in Proteomic Analysis. Proteomics 2014, 14, 645–658.
- Nishii, K.; Möller, M.; Foster, R.G.; Forrest, L.L.; Kelso, N.; Barber, S.; Howard, C.; Hart, M.L. A High Quality, High Molecular Weight DNA Extraction Method for PacBio HiFi Genome Sequencing of Recalcitrant Plants. Plant Methods 2023, 19, 41.
- Barbier, F.F.; Chabikwa, T.G.; Ahsan, M.U.; Cook, S.E.; Powell, R.; Tanurdzic, M.; Beveridge, C.A. A Phenol/Chloroform-Free Method to Extract Nucleic Acids from Recalcitrant, Woody Tropical Species for Gene Expression and Sequencing. Plant Methods 2019, 15, 62.
- Valizadeh, N.; Holasou, H.A.; Mohammadi, S.A.; Khawar, K.M. A Comparison of Genomic DNA Extraction Protocols in Artemisia Annua L. for Large Scale Genetic Analyses Studies. Iran. J. Sci. Technol. Trans. Sci. 2021, 45, 1587–1595.
- Springer, N.M. Isolation of Plant DNA for PCR and Genotyping Using Organic Extraction and CTAB. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5515.
- Spadoni, A.; Sion, S.; Gadaleta, S.; Savoia, M.A.; Piarulli, L.; Fanelli, V.; di Rienzo, V.; Taranto, F.; Miazzi, M.; Montemurro, C.; et al. A Simple and Rapid Method for Genomic DNA Extraction and Microsatellite Analysis in Tree Plants. J. Agric. Sci. Technol. 2019, 21, 1215–1226.
- Koh, R.B.L.; Barbosa, C.F.C.; Aquino, V.M.; Galvez, L.C. Extraction of High Molecular Weight DNA Suitable for Next-Generation Sequencing from the Fiber Crop Abaca. Ind. Crops Prod. 2021, 161, 113194.
- Ali, Q.; Salisu, I.B.; Raza, A.; Shahid, A.A.; Rao, A.Q.; Husnain, T. A Modified Protocol for Rapid DNA Isolation from Cotton (Gossypium spp.). Methods X 2019, 6, 259–264.
- Lee, B.-J.; Kim, S.; Lee, J.-W.; Lee, H.-M.; Eo, S.H. Technical Note: Polyvinylpyrrolidone (PVP) and Proteinase-K Improve the Efficiency of DNA Extraction from Japanese Larch Wood and PCR Success Rate. Forensic Sci. Int. 2021, 328, 111005.
- Brandão, W.Q.; da Silva, R.J.; Mojica-Sánchez, L.C.; Maciel, B.G.; Ratkovski, G.P.; de Melo, C.P. Use of Polypyrrole-Polystyrene Membranes for Extracting DNA from Plant Tissues. Biomater. Biosyst. 2022, 7, 100060.
- Natarajan, V.P.; Zhang, X.; Morono, Y.; Inagaki, F.; Wang, F. A Modified SDS-Based DNA Extraction Method for High Quality Environmental DNA from Seafloor Environments. Front. Microbiol. 2016, 7, 986.
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A Modified Protocol for Rapid DNA Isolation from Plant Tissues Using Cetyltrimethylammonium Bromide. Nat. Protoc. 2006, 1, 2320–2325.
- Jiang, Q.; Li, Y.; Huang, L.; Guo, J.; Wang, A.; Ma, C.; Shi, C. Direct Capture and Amplification of Nucleic Acids Using a Universal, Elution-Free Magnetic Bead-Based Method for Rapid Pathogen Detection in Multiple Types of Biological Samples. Anal. Bioanal. Chem. 2023, 415, 427–438.
- Oberacker, P.; Stepper, P.; Bond, D.M.; Hoehn, S.; Focken, J.; Meyer, V.; Schelle, L.; Sugrue, V.J.; Jeunen, G.-J.; Moser, T.; et al. Bio-On-Magnetic-Beads (BOMB): Open Platform for High-Throughput Nucleic Acid Extraction and Manipulation. PLoS Biol. 2019, 17, e3000107.
- Fort, A.; Guiry, M.D.; Sulpice, R. Magnetic Beads, a Particularly Effective Novel Method for Extraction of NGS-Ready DNA from Macroalgae. Algal Res. 2018, 32, 308–313.
- Fei, Z.; Cheng, C.; Wei, R.; Tan, G.; Xiao, P. Reversible Superhydrophobicity Unyielding Magnetic Beads of Flipping-Triggered (SYMBOL) Regulate the Binding and Unbinding of Nucleic Acids for Ultra-Sensitive Detection. Chem. Eng. J. 2022, 431, 133953.
- Balakrishnan, S.G.; Ahmad, M.R.; Koloor, S.S.R.; Petrů, M. Separation of CtDNA by Superparamagnetic Bead Particles in Microfluidic Platform for Early Cancer Detection. J. Adv. Res. 2021, 33, 109–116.
- Hin, S.; Paust, N.; Rombach, M.; Lueddecke, J.; Specht, M.; Zengerle, R.; Mitsakakis, K. Magnetophoresis in Centrifugal Microfluidics at Continuous Rotation for Nucleic Acid Extraction. Micromachines 2022, 13, 2112.
- Wu, J.; Kodzius, R.; Cao, W.; Wen, W. Extraction, Amplification and Detection of DNA in Microfluidic Chip-Based Assays. Microchim. Acta 2014, 181, 1611–1631.
- Tian, W.; Liu, Y.; Wang, S.; Ye, J.; Liu, H.; Wang, Y.; Zhou, M. Automated and Rapid Easy-to-Use Magnetic Solid-Phase Extraction System for Five Heavy Metals in Cereals and Feeds. Foods 2022, 11, 3944.
- Tong, R.; Zhang, L.; Hu, C.; Chen, X.; Song, Q.; Lou, K.; Tang, X.; Chen, Y.; Gong, X.; Gao, Y.; et al. An Automated and Miniaturized Rotating-Disk Device for Rapid Nucleic Acid Extraction. Micromachines 2019, 10, 204.
- Li, J.; Gao, H.; Li, Y.; Xiao, F.; Zhai, S.; Wu, G.; Wu, Y. Event-Specific PCR Methods to Quantify the Genetically Modified DBN9936 Maize. J. Food Compos. Anal. 2022, 105, 104236.
- Spanea, E.; Tsironi, T.; Tsakali, E.; Batrinou, A.; Stefanou, V.; Antonopoulos, D.; Koussissis, S.; Tsaknis, J.; Impe, J.V.; Houhoula, D. Evaluation of a Real Time PCR Assay Method for the Detection of Genetically Modified Organisms in Food Products. J. Food Res. 2020, 9, 1.
- Kaur, J.; Radu, S.; Ghazali, F.M.; Kqueen, C.Y. Real-Time PCR-Based Detection and Quantification of Genetically Modified Maize in Processed Feeds Commercialised in Malaysia. Food Control 2010, 21, 1536–1544.
- Dong, S.; Zhang, D.; Yu, C.; Zhang, Z.; Liu, Y. Using Droplet Digital PCR to Detect Plant DNA in Tissues of Zebrafish (Danio Rerio) Fed Genetically Modified Maize. Aquac. Res. 2021, 52, 4467–4474.
- Zimmermann, A.; Hemmer, W.; Liniger, M.; Luthy, J.; Pauli, U. A Sensitive Detection Method for Genetically Modified MaisGard (TM) Corn Using a Nested PCR-System. Food Sci. Technol.-Lebensm.-Wiss. Technol. 1998, 31, 664–667.
- Dong, L.; Long, L.; Xing, Z.; Li, C.; He, Y.; Yan, W.; Xia, W.; Li, F. Establishment of Multi-Fluorescence Real-Time PCR Assay for GM Soybean Detection. Int. J. Agric. Biol. 2020, 24, 292–298.
- Godbey, W.T. Chapter 10—The Polymerase Chain Reaction (PCR). In Biotechnology and its Applications, 2nd ed.; Godbey, W.T., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 219–246. ISBN 978-0-12-817726-6.
- Stephenson, F.H. Chapter 8—The Polymerase Chain Reaction. In Calculations for Molecular Biology and Biotechnology, 3rd ed.; Stephenson, F.H., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 171–213. ISBN 978-0-12-802211-5.
- Dobnik, D.; Spilsberg, B.; Bogožalec Košir, A.; Štebih, D.; Morisset, D.; Holst-Jensen, A.; Žel, J. Multiplex Droplet Digital PCR Protocols for Quantification of GM Maize Events. Methods Mol. Biol. Clifton NJ 2018, 1768, 69–98.
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real Time Quantitative PCR. Genome Res. 1996, 6, 986–994.
- Fengjun, W.; Qiang, C.; Sudan, Y.; Haifeng, X.; Hongxia, X.; Yiping, Y.; Yexi, Z. Rapid Identification of Transgenic Maize Lines and Transformants by Quadruplex Real-Time PCR. Zhongguo Liangyou Xuebao 2021, 36, 128–135.
- McNutt, M.K.; Baulcombe, D.; Benfey, P.N.; Bergmann, D.; Coruzzi, G.M.; Estelle, M.; Kieber, J.J.; Long, S.P.; Niyogi, K.K.; Schroeder, J.I.; et al. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241.
- Tellinghuisen, J. DPCR vs. QPCR: The Role of Poisson Statistics at Low Concentrations. Anal. Biochem. 2020, 611, 113946.
- Chen, S.; Sun, Y.; Fan, F.; Chen, S.; Zhang, Y.; Zhang, Y.; Meng, X.; Lin, J.-M. Present Status of Microfluidic PCR Chip in Nucleic Acid Detection and Future Perspective. TrAC Trends Anal. Chem. 2022, 157, 116737.
- Bogožalec Košir, A.; Demšar, T.; Štebih, D.; Žel, J.; Milavec, M. Digital PCR as an Effective Tool for GMO Quantification in Complex Matrices. Food Chem. 2019, 294, 73–78.
- Dong, L.; Meng, Y.; Sui, Z.; Wang, J.; Wu, L.; Fu, B. Comparison of Four Digital PCR Platforms for Accurate Quantification of DNA Copy Number of a Certified Plasmid DNA Reference Material. Sci. Rep. 2015, 5, 13174.
- Wan, J.; Song, L.; Wu, Y.; Brzoska, P.; Keys, D.; Chen, C.; Valliyodan, B.; Shannon, J.G.; Nguyen, H.T. Application of Digital PCR in the Analysis of Transgenic Soybean Plants. Adv. Biosci. Biotechnol. 2016, 7, 403–417.
- Demeke, T.; Dobnik, D. Critical Assessment of Digital PCR for the Detection and Quantification of Genetically Modified Organisms. Anal. Bioanal. Chem. 2018, 410, 4039–4050.
- Kahkeshani, S.; Kong, J.E.; Wei, Q.; Tseng, D.; Garner, O.B.; Ozcan, A.; Di Carlo, D. Ferrodrop Dose-Optimized Digital Quantification of Biomolecules in Low-Volume Samples. Anal. Chem. 2018, 90, 8881–8888.
- Park, J.; Han, D.H.; Hwang, S.-H.; Park, J.-K. Reciprocating Flow-Assisted Nucleic Acid Purification Using a Finger-Actuated Microfluidic Device. Lab. Chip 2020, 20, 3346–3353.
- Park, J.; Lee, K.G.; Han, D.H.; Lee, J.-S.; Lee, S.J.; Park, J.-K. Pushbutton-Activated Microfluidic Dropenser for Droplet Digital PCR. Biosens. Bioelectron. 2021, 181, 113159.
- Cottenet, G.; Blancpain, C.; Chuah, P.F. Performance Assessment of Digital PCR for the Quantification of GM-Maize and GM-Soya Events. Anal. Bioanal. Chem. 2019, 411, 2461–2469.
- Shen, C.; Yin, H.; Tong, Z.; Qiu, S.; Lu, Y.; Wu, Z.; Mao, H. Digital Microfluidic Chip Based on Direct Ink Writing For Nucleic Acid Multiplex PCR Detection. In Proceedings of the 2022 IEEE 35th International Conference on Micro Electro Mechanical Systems Conference (MEMS), Tokyo, Japan, 9–13 January 2022; pp. 365–368.
- Shin, M.K.; Jeon, S.M.; Koo, Y.E. Development of a Rapid Detection Method for Genetically Modified Rice Using the Ultra-Fast PCR System. Food Sci. Biotechnol. 2022, 31, 175–182.
- Zhou, H.; Lei, Y.; Wang, P.; Liu, M.; Hu, X. Development of SYBR Green Real-Time PCR and Nested RT-PCR for the Detection of Potato Mop-Top Virus (PMTV) and Viral Surveys in Progeny Tubers Derived from PMTV Infected Potato Tubers. Mol. Cell. Probes 2019, 47, 101438.
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, E63.
- Lizardi, P.M.; Huang, X.; Zhu, Z.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation Detection and Single-Molecule Counting Using Isothermal Rolling-Circle Amplification. Nat. Genet. 1998, 19, 225–232.
- Xu, G.; Hu, L.; Zhong, H.; Wang, H.; Yusa, S.; Weiss, T.C.; Romaniuk, P.J.; Pickerill, S.; You, Q. Cross Priming Amplification: Mechanism and Optimization for Isothermal DNA Amplification. Sci. Rep. 2012, 2, 246.
- Compton, J. Nucleic-Acid Sequence-Based Amplification. Nature 1991, 350, 91–92.
- Lasken, R.S. Genomic DNA Amplification by the Multiple Displacement Amplification (MDA) Method. Biochem. Soc. Trans. 2009, 37, 450–453.
- Vincent, M.; Xu, Y.; Kong, H.M. Helicase-Dependent Isothermal DNA Amplification. Embo Rep. 2004, 5, 795–800.
- Lobato, I.M.; O’Sullivan, C.K. Recombinase Polymerase Amplification: Basics, Applications and Recent Advances. TrAC Trends Anal. Chem. 2018, 98, 19–35.
- Chalmers, F.M.; Curnow, K.M. Scaling Up the Ligase Chain Reaction-Based Approach to Gene Synthesis. BioTechniques 2001, 30, 249–252.
- Ehses, S.; Ackermann, J.; McCaskill, J.S. Optimization and Design of Oligonucleotide Setup for Strand Displacement Amplification. J. Biochem. Biophys. Methods 2005, 63, 170–186.
- Toley, B.J.; Covelli, I.; Belousov, Y.; Ramachandran, S.; Kline, E.; Scarr, N.; Vermeulen, N.; Mahoney, W.; Lutz, B.R.; Yager, P. Isothermal Strand Displacement Amplification (ISDA): A Rapid and Sensitive Method of Nucleic Acid Amplification for Point-of-Care Diagnosis. Analyst 2015, 140, 7540–7549.
- Singh, M.; Pal, D.; Sood, P.; Randhawa, G. Loop-Mediated Isothermal Amplification Assays: Rapid and Efficient Diagnostics for Genetically Modified Crops. Food Control 2019, 106, 106759.
- Xu, J.; Zheng, Q.; Yu, L.; Liu, R.; Zhao, X.; Wang, G.; Wang, Q.; Cao, J. Loop-Mediated Isothermal Amplification (LAMP) Method for Detection of Genetically Modified Maize T25. Food Sci. Nutr. 2013, 1, 432–438.
- Li, R.; Chen, J.; Zhang, X.; Cui, J.; Tao, S.; Yang, L. Mini-Disk Capillary Array Coupling with LAMP for Visual Detection of Multiple Nucleic Acids Using Genetically Modified Organism Analysis as an Example. J. Agric. Food Chem. 2020, 68, 899–906.
- Kaymaz, S.V.; Elitas, M. Optimization of Loop-Mediated Isothermal Amplification (LAMP) Reaction Mixture for Biosensor Applications. MethodsX 2021, 8, 101282.
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204.
- Choi, G.; Jung, J.H.; Park, B.H.; Oh, S.J.; Seo, J.H.; Choi, J.S.; Kim, D.H.; Seo, T.S. A Centrifugal Direct Recombinase Polymerase Amplification (Direct-RPA) Microdevice for Multiplex and Real-Time Identification of Food Poisoning Bacteria. Lab. Chip 2016, 16, 2309–2316.
- Chandu, D.; Paul, S.; Parker, M.; Dudin, Y.; King-Sitzes, J.; Perez, T.; Mittanck, D.W.; Shah, M.; Glenn, K.C.; Piepenburg, O. Development of a Rapid Point-of-Use DNA Test for the Screening of Genuity® Roundup Ready 2 Yield® Soybean in Seed Samples. BioMed Res. Int. 2016, 2016, 3145921.
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958.
- Versatile Isothermal DNA/RNA Amplification by TwistDx. Available online: https://www.twistdx.co.uk/ (accessed on 17 July 2023).
- Lin, K.; Guo, J.; Guo, X.; Li, Q.; Li, X.; Sun, Z.; Zhao, Z.; Weng, J.; Wu, J.; Zhang, R.; et al. Fast and Visual Detection of Nucleic Acids Using a One-Step RPA-CRISPR Detection (ORCD) System Unrestricted by the PAM. Anal. Chim. Acta 2023, 1248, 340938.
- Li, K.; Luo, Y.; Huang, K.; Yang, Z.; Wan, Y.; Xu, W. Single Universal Primer Recombinase Polymerase Amplification-Based Lateral Flow Biosensor (SUP-RPA-LFB) for Multiplex Detection of Genetically Modified Maize. Anal. Chim. Acta 2020, 1127, 217–224.
- Liu, R.; Wang, Z.; Liu, X.; Chen, A.; Yang, S. Rapid On-Site Detection of Salmonella Pullorum Based on Lateral Flow Nucleic Acid Assay Combined with Recombinase Polymerase Amplification Reaction. Poult. Sci. 2020, 99, 7225–7232.
- Seder, I.; Coronel-Tellez, R.; Helalat, S.H.; Sun, Y. Fully Integrated Sample-in-Answer-out Platform for Viral Detection Using Digital Reverse Transcription Recombinase Polymerase Amplification (DRT-RPA). Biosens. Bioelectron. 2023, 237, 115487.
- Tortajada-Genaro, L.A.; Maquieira, A. Multiple Recombinase Polymerase Amplification and Low-Cost Array Technology for the Screening of Genetically Modified Organisms. J. Food Compos. Anal. 2021, 103, 104083.
- Glökler, J.; Lim, T.S.; Ida, J.; Frohme, M. Isothermal Amplifications—A Comprehensive Review on Current Methods. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 543–586.
- Wang, Y.; Sun, L.; Li, J.; Wang, Z.; Jiao, W.; Xiao, J.; Shen, C.; Xu, F.; Qi, H.; Wang, Y.; et al. Label-Free Cross-Priming Amplification Coupled with Endonuclease Restriction and Nanoparticles-Based Biosensor for Simultaneous Detection of Nucleic Acids and Prevention of Carryover Contamination. Front. Chem. 2019, 7, 322.
- Nguyen, H.Q.; Nguyen, V.D.; Van Nguyen, H.; Seo, T.S. Quantification of Colorimetric Isothermal Amplification on the Smartphone and Its Open-Source App for Point-of-Care Pathogen Detection. Sci. Rep. 2020, 10, 15123.
- Turkec, A.; Lucas, S.J.; Karacanli, B.; Baykut, A.; Yuksel, H. Assessment of a Direct Hybridization Microarray Strategy for Comprehensive Monitoring of Genetically Modified Organisms (GMOs). Food Chem. 2016, 194, 399–409.
- Wei, B.-R.; Savellano, D.F.; Hu, C.-H. Analysis of Status Quo and Research Progress in Nursing Care for Different Typed Coronavirus Disease 2019. J. Integr. Nurs. 2020, 2, 160–171.
- Morisset, D.; Dobnik, D.; Hamels, S.; Zel, J.; Gruden, K. NAIMA: Target Amplification Strategy Allowing Quantitative on-Chip Detection of GMOs. Nucleic Acids Res. 2008, 36, e118.
- Ki, E.Y.; Lee, Y.K.; Lee, A.; Park, J.S. Comparison of the PANArray HPV Genotyping Chip Test with the Cobas 4800 HPV and Hybrid Capture 2 Tests for Detection of HPV in ASCUS Women. Yonsei Med. J. 2018, 59, 662–668.
- Lu, X.-B.; Wu, H.-B.; Wang, M.; Li, B.-D.; Yang, C.-L.; Sun, H.-W. Developing a Method of Oligonucleotide Microarray for Event Specific Detec-Tion of Transgenic Maize. ACTA Agron. Sin. 2009, 35, 1432–1438.
- Liu-Stratton, Y.; Roy, S.; Sen, C.K. DNA Microarray Technology in Nutraceutical and Food Safety. Toxicol. Lett. 2004, 150, 29–42.
- Gao, H.; Yan, C.; Wu, W.; Li, J. Application of Microfluidic Chip Technology in Food Safety Sensing. Sensors 2020, 20, 1792.
This entry is offline, you can click here to edit this entry!