Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Exercise produces oxidants from a variety of intracellular sources, including NADPH oxidases (NOX) and mitochondria. Exercise-derived reactive oxygen species (ROS) are beneficial, and the amount and location of these ROS is important to avoid muscle damage associated with oxidative stress.
- ATP production
- mitochondrial network
- mitochondria dynamics
1. Introduction
Since the first report that muscular physiology is redox-dependent, several research lines in redox biology have grown significantly. Skeletal muscle is the human buffer against aging, some metabolism pathologies, and neuropathologies, so the understanding of how skeletal physiology processes are modulated by reactive oxygen species (ROS) is a source of potential intervention based on diet supplementation or exercise protocols. Evidence suggests that exercise produces oxidants from a variety of intracellular sources, including NADPH oxidases (NOX) and mitochondria as the main sources [1,2]. Exercise-derived ROS are beneficial, and the amount and location of these ROS is important to avoid muscle damage associated with oxidative stress.
2. An Energy-Saving Mechanism in Skeletal Muscle
2.1. Mitochondria Distribution and Characteristics in Skeletal Muscle
Skeletal muscle fibers can change from a preferentially glycolytic metabolism to oxidative metabolism upon certain types of physiological exercise, a process for which the cellular mechanism needs to be better understood. In turn, the excitation–contraction (EC) mechanism is connected to a network of mitochondria in skeletal muscle fibers, showing a physiological communication between contractile machinery and mitochondria, in which Ca2+ has a relevant role. Moreover, mitochondria are mobile and plastic organelles, constantly changing shape, fusing, or fissioning with each other, and changing their role in parallel in cellular bioenergetics [3].
Adult skeletal muscle presents two populations of mitochondria: one group comprises both perinuclear (PN) and perivascular (PV) mitochondria, both considered peripherally located mitochondria (PLM) or subsarcolemmal mitochondria (SSM); the other group is called intermyofibrillar (IMF) mitochondria (see Figure 1) [4]. The SSM population comprises the mitochondria located beneath the plasma membrane of the muscle fiber (and, as nuclei have a similar location in adult skeletal muscle, mitochondria closely surrounding the myonuclei) and the IMF population of mitochondria, located regularly close to the sarcoplasmic reticulum terminal cisternae and the triad (the grouping of two terminal cisternae an one transverse tubule), at regular intervals along every myofibril [5,6].
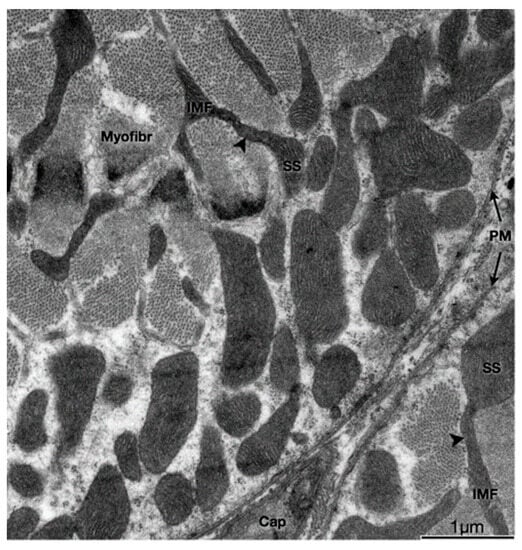
Figure 1. Physical interactions between SS and IMF mitochondria. Transmission electron micrograph of myofibers in the transverse plane. SS and IMF mitochondria are distinct organelles. Some SS and IMF mitochondria form continuous organelles (arrowheads) that coexist in both subcellular compartments. SS, Subsarcolemmal; IMF, intermyofibrillar; PM, plasma membrane (sarcolemma); Myofibr, myofibrils; Cap, capillary.
IMF mitochondria form a structural arrangement characterized by the interaction of transverse mitochondrial tubules in the sarcomere, called “mitochondrial reticulum”, which has been proposed as an energetic conductive pathway from mitochondria to the contractile apparatus [4]. Specialized proteins play roles such as intermembrane linkers, which are formed by a single protein with two membrane-interacting domains in the sarcoplasmic reticulum [8]. This physical proximity, such as voltage-dependent anion channel (VDAC) and RyR1 or IP3R, allows the passage of calcium from the reticulum to the mitochondria to be more effective. It has also been described that H2O2 would diffuse from the mitochondrial space towards these contact domains to modulate Ca2+ release locally [8,9]. It is very interesting to note that these two populations of mitochondria with different membrane potential not only contribute both to muscle fiber oxidative capacity and bioenergetics but also do so each in a specialized way, allowing in the end, to optimize ATP production precisely where is needed for contraction, i.e., near the myofibrils, by producing mitochondria membrane potential propagation. In agreement with this, it has been proposed that SSM supports the IMF energy, based on the presence of higher oxidative enzyme activity [10].
Together with the different localization and oxidative capacity, both populations express different types of proteins and different membrane potentials. For example, SSM expresses mitochondrial calcium uniporter regulator 1 (MICU1) [11]. Mitochondrial calcium uniporter (MCU) is a highly selective and highly regulated calcium channel inserted in the inner mitochondrial membrane that allows calcium uptake from the cytosol to mitochondria after contraction [10,12]. This complex (when regulated by MICU1) is basally closed and is activated upon cytoplasmic Ca2+ increases, allowing then the ion to enter the mitochondria matrix. ROS production is normally high when the mitochondrial potential (ΔΨ) is elevated. Therefore, MICU1 expression, by regulating Ca2+ entry, would protect against mitochondrial H2O2-generated damage [13].
This idea is consistent with the evidence showing that, in neurons, MCU promoted the activity of the electron transport chain and the chemical reduction of NAD+ to NADH, which would imply that electrons are not available for ROS formation [14]. Moreover, it is important to highlight that high MCU activity also induces mitochondrial membrane potential dissipation, facilitating the activity of the ATP synthase [15]. It has been observed that the lower expression of MCU generates muscle atrophy, intimately relating the regulation of mitochondrial Ca2+ to the size of the muscle fiber [16]. The action of MCU is crucial for ATP synthesis in aerobic conditions because Ca2+ increase is an essential cofactor for the tricarboxylic acid (TCA) cycle’s enzymes such as glycerol phosphate dehydrogenase (GPDH), pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH) [17]. Thus, Ca2+ increase can elevate the efficiency of complexes I, III, and IV of OXPHOS. The Ca2+-induced activation of mitochondrial sodium–calcium exchanger (NCLX) results in a Na+ influx into the matrix, and Na+ interacts with phospholipids in the inner leaflet of the inner membrane, decreases its fluidity, and slows down ubiquinol (UQH2) diffusion, increasing electron flux due to major NADH availability. In simple words, calcium potentiates the establishment of the proton gradient needed for ATP synthesis, which is used for sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) to pump back calcium from the cytosol to the SR [18].
When we talk about muscle activity, it is important to remember that there are different types of muscle fibers (as different types of motor units). Slow, fatigue-resistant fibers possess a larger number of mitochondria, having a preferential oxidative metabolism that allows them to contract for long periods of time. Fast, fatigable fibers have lower mitochondria content, relying on an anaerobic glycolytic metabolism, which is responsible for their poor fatigue resistance. These different types of muscle fibers can change from one phenotype to another depending on external demands. One of the main actors in maintaining or changing the muscle phenotype is the pattern of stimulation coming from motor neurons. In this way, it has been shown that low-frequency electrical stimulation induces the expression of genes belonging to the slow phenotype, while high frequencies lead to the expression of genes typical for fast phenotypes. Among the slow phenotype transcriptional profile, we can find slow isoforms of proteins from the contractile apparatus and of factors that lead to increased mitochondrial biogenesis as well as oxidative enzymes [19,20]. Interestingly, Quezada et al. have also shown that low-frequency electrical stimulation (which allows fibers to convert to slow-phenotype fibers) can decrease the expression of the MCU complex in isolated adult fibers [21].
Higher protein levels of MCU and MICU1 per mitochondrion have been observed in fast-phenotypes muscles (like flexor digitorium longus) than in slow phenotype muscles, such as the soleus. From these data, we can propose a hypothesis in which the decrease in mRNA of the MCU complex after the low-frequency electrical stimulation of isolated fibers from a fast muscle would favor a lower protein level of MCU and MICU1 per mitochondrion, being an early metabolic response to the phenotypic shift from fast to slow phenotype muscle fiber [22]. Also, a gradual increase in the number of mitochondria, together with a decrease in levels of the MCU complex in response to a low-frequency electrical stimulus, could allow adapting mitochondrial Ca2+ homeostasis to finally reach that of a slow muscle. On the other hand, Mcu gen deletion produces a decrease in Ca2+-stimulated ATP synthesis, impairment in TCA cycle substrate flux, and a turn toward fatty acid metabolism [12].
2.2. Function of the Heterogeneity of Mitochondria within the Muscle Fiber
The SS mitochondria are quite different to the IMF mitochondria [7,11,23]. It has been shown that electron transport chain elements needed to establish the mitochondria membrane potential are differentially distributed among SSM and IMF; in particular, complex IV and cytochrome C are located mostly in the SSM [11,23]. There are also differences in the distribution of complex V (ATP synthase), which is located mostly in the IMF mitochondria [23]. In contrast, the importin translocase of the outer membrane, TOM20, has homogeneous distribution in all the cell’s mitochondria, suggesting a specific compartmentalization of different mitochondrial proteins between mitochondria subpopulations [11].
This evidence, together with the fact that there is a shift in mitochondria membrane potential (higher in SSM in resting conditions) towards the center of the muscle fiber upon electrical stimulation [11], prompted us to propose that, in the SSM pool, the main proton motive force is generated by the activation of complexes I-IV. In contrast, ATP is generated in complex V, which is in the IMF mitochondria, and this ATP production will occur only after the fusion of both mitochondria populations with the consequent spreading of mitochondria membrane potential. As mitochondria membrane potential is established across the inner mitochondria membrane, it can only propagate via the fusion of mitochondria membranes. Mitochondrial ROS production will be minimal since the proton motive force is used for ATP synthesis when the electron transport chain works at a maximum level induced by exercise.
In fact, Mcu deletion produces a decrease in Ca2+-stimulated ATP synthesis, impairment in TCA cycle substrate flux, and a turn toward fatty acid metabolism [12]. Stimuli, such as extracellular ATP or electrical stimulation, can increase the expression of MCU in isolated adult fibers [21]. In turn, it has been shown that the MCU-dependent increase in mitochondrial ROS is necessary for optimal skeletal muscle repair after an injury via the induction of actin polymerization dependent on RhoA [24]. It is interesting to note that, using the direct measurement of superoxide via electron paramagnetic resonance, Crochemore et al. demonstrated that SSM produce more superoxide than IMF mitochondria [25].
In summary, skeletal muscle mitochondria achieve this performance by separating two important functions: the first is mitochondria membrane potential generation via calcium-sensitive oxidative phosphorylation (which occurs mainly in the subsarcolemmal population of mitochondria), and the second function is ATP production mediated by ATP synthase, which occurs in the intermyofibrillar population of mitochondria. To achieve this amazing performance, the two populations of mitochondria are not connected at rest and have a different resting protein composition. The intermyofibrillar mitochondria are enriched in ATP synthase and have a high content of mitochondrial calcium uptake 1 (MICU1), MICU1, as a key regulator of mitochondrial Ca2+ uptake, which negatively regulates calcium entry into the mitochondrial matrix through the MCU calcium channel [26,27]. This protein is a Ca2+ sensor, and its functioning does not impact ΔΨ or oxygen consumption [26,28]. On the other hand, the subsarcolemmal mitochondria are enriched in the electron transport chain complex proteins and, having no MICU1, can reach a high calcium content upon muscle activation (Figure 2). The model in Figure 2 shows that exercise induces the mitochondrial fusion of SSM and IMF, transferring electrical properties and proteins from SSM to the mitochondrial network, decreasing the ROS generation, and improving energy-saving design. This process is reversible, and, consequently, in resting conditions after exercise, the fission of mitochondria will occur, and the high mitochondria membrane potential in SSM can be recovered.
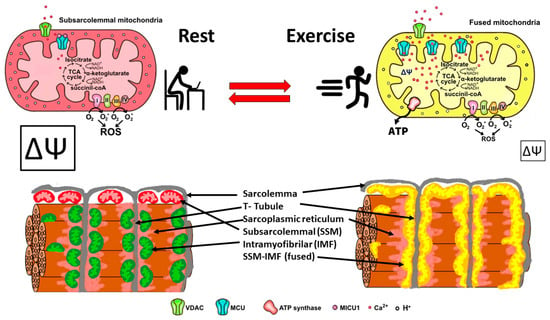
Figure 2. Energy efficient distribution of mitochondria that saves energy and minimizes mitochondria ROS production. Under resting conditions, skeletal muscle has different pools of mitochondria, SSM have a high mitochondrial potential (ΔΨ) and a basal rate of ROS generation from superoxide anions generated by the electron transport chain. Thus, the respiratory chain may allow the “leakage of electrons” and generate ROS. IMF mitochondria have a high expression of MICU1, which prevents calcium from massively entering the mitochondrial space. A condition of exercise implies that the SSM and the IMF mitochondria fuse, generating a network characterized by a lower density of MICU1, a high density of MCU, and an extended proton gradient (enough ΔΨ) needed to generate ATP and to minimize ROS generation. Colors of mitochondria represent diversity (red and green), and yellow represents fused mitochondria.
2.3. Mitochondria Dynamics Are Altered in Skeletal Muscle of Aging Subjects and in Pathological Conditions
Mitochondrial fusion events in skeletal muscle fibers are hard to evidence due to the highly restricted space in which IMF mitochondria are located; fusion events nevertheless take place, and they were shown for the first time by Eisner et al. in 2014 [29].
When we consider the above-described model, it is reasonable to assume that mitochondria dynamics (fission and fusion) play an essential role in skeletal muscle function and wellbeing. Maintaining optimal skeletal muscle health requires dynamic mitochondrial function, which becomes disrupted in aging and various pathological conditions. Age-related mitochondrial dysfunction is characterized by impaired fusion/fission processes and mitophagy, leading to sarcopenia and reduced exercise capacity. In addition, diseases such as muscular dystrophies, mitochondrial myopathies, and type 2 diabetes exhibit mitochondrial fusion and fission imbalances, contributing to impaired excitation–transcription coupling (ETC, see Figure 3).
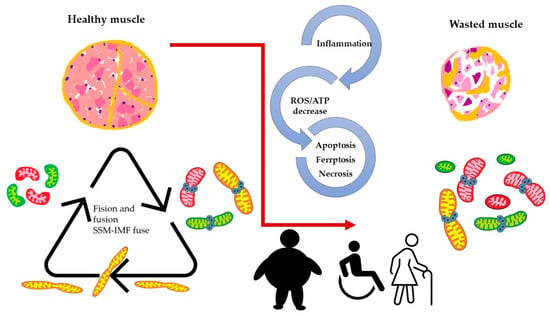
Figure 3. Altered dynamics and fusion capacity of mitochondria in disease. A common factor in many pathological conditions affecting skeletal muscle is an imbalance in mitochondrial dynamics. This imbalance is both a cause and consequence of inflammation, increased ROS production, and cell death, leading to muscle deterioration characterized by dysfunctional mitochondria. Different colors of mitochondria refer to SSM, IFM and fused mitochondria as pictures in Figure 2.
Alterations in mitochondria dynamics have been shown to occur in middle age and more advanced age in mice [30], and they parallel dramatic decreases in muscle function. This was evidenced by changes in mitochondria orientation as seen by confocal microscopy, changes in both mitochondria size and shape as seen by electron microscopy, and changes in the expression of proteins involved in fission and fusion processes. Furthermore, mitochondria dynamics appeared to be altered in the skeletal muscle of a mouse model of alcohol consumption [29]. Oxidative stress and reduced antioxidants reinforce mitochondrial fragmentation, suppress fusion/fission, and impair the electron transport chain, decrease ATP production, and cause DNA damage. Mitochondrial dynamics depend on proteins such as mitofusin (MFN) 1 and 2 and optic atrophy protein 1(OPA1) for the fusion of the outer and inner mitochondrial membranes. Mitochondrial fission involves the recruitment of cytoplasmic Drp1 to the mitochondrial outer membrane, forming a ring-like structure with adaptors Mff, MiD49, and MiD51. As schematized in Figure 3, these protein interactions ensure a balance between fusion and fission, which is crucial for maintaining mitochondrial dynamics and cellular functions [31]. The deletion of OPA1 leads to mitochondrial dysfunction, reduced myogenic stem cells, decreased protein synthesis, and the activation of protein breakdown. Opa1 deletion in skeletal muscle affects the entire body, causing a premature aging phenotype that ultimately leads to animal death [32]. Opa1 deficiency in myopathy engages TLR9, activating NF-κB and triggering muscle inflammation. This localized inflammation can become systemic, impacting the entire body. An altered growth hormone/IGF1 axis and enhanced FGF21 expression are observed, contributing to impaired growth. Opa1 deficiency disrupts growth-related processes and promotes inflammation via inflammatory pathway activation. These processes collectively contribute to the development and progression of myopathy [33]. Aging is associated with sarcopenia, and when obesity is associated, the term sarcopenic obesity (SO) is applied. Dysfunctional adipose tissue, fatty acid excess inside the bloodstream, and low-grade systemic inflammation are combined, resulting in lipotoxicity, oxidative stress, insulin resistance, and inflammation in the skeletal muscle [34]. It has been reported that a high-fat diet suppresses mitochondrial biogenesis in the skeletal muscle of zebrafish and decreases the number of SSM, Mfn2, and OPA1. On the contrary, Fis1 and Drp1, both fission proteins, were found to be increased in an obese mouse model, as opposed to in non-obese conditions in mice [31]. On the other hand, mitochondrial uncoupling attenuates SO by enhancing skeletal muscle mitophagy, reducing muscle inflammation, promoting mitochondrial turnover via STAT3 signaling, and mitigating muscle degradation [35].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12081624
This entry is offline, you can click here to edit this entry!