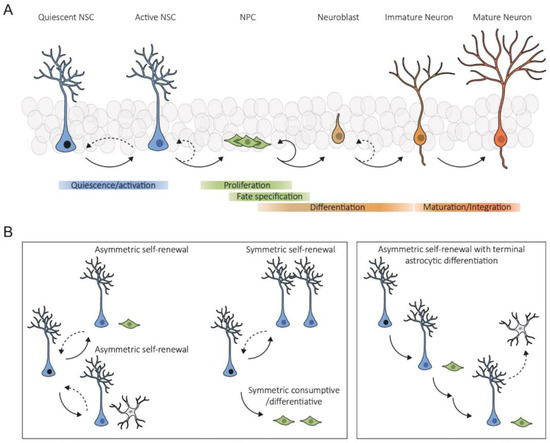
2. Intrinsic Mechanisms
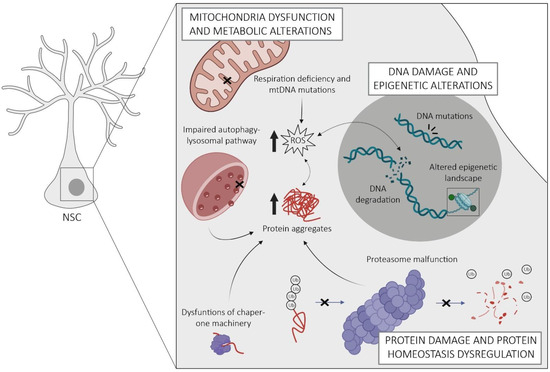
The regulation of NSCs is inherently complex and crucial for maintaining their regenerative potential throughout life. Within these cells, there exist intricate and interconnected mechanisms that homeostatically regulate their molecular, structural, and functional properties, which become dysregulated with age, adversely affecting the functions of NSCs. Researchers provide an overview of the currently known intrinsic mechanisms underlying NSC aging, encompassing alterations in mitochondrial function and metabolism, disturbances in protein homeostasis, DNA damage, and alterations in epigenetic patterns, as well as the onset of cellular senescence (Figure 2).
Figure 2. Schematic representation of the key intrinsic mechanisms of NSC aging in the SGZ. Aged NSCs are characterized by mitochondrial dysfunction leading to the accumulation of reactive oxygen species (ROS) and oxidative stress. This causes damage to cellular components such as proteins and DNA, which entails the formation of protein aggregates and the accumulation of DNA mutations. Despite this, aged NSCs exhibit a dysregulated protein homeostasis arising from malfunctions of the proteasome, chaperones, and autophagy–lysosomal pathways. Changes in their epigenetic landscape lead to alterations in gene expression patterns. Notes: the non-dashed and dashed arrows indicate unidirectional and bidirectional relationships, respectively. Crosses indicate a malfunction.
2.1. Protein Homeostasis Dysregulation
The accumulation of misfolded or damaged proteins is a critical determinant of stem cell proliferation and aging [
28]. Similar to other stem cells, adult NSCs use multiple interconnected pathways to regulate the balance of intracellular proteins (proteostasis), including molecular chaperones, the autophagy–lysosomal pathway, and the proteasome. Chaperones play a central role at all levels of proteostasis by facilitating proper folding of nascent or damaged proteins, assisting in protein translocation and stabilization, preventing protein aggregation, and supporting protein degradation via the ubiquitin–proteasome system and autophagy [
28,
29]. The proteasome is responsible for the degradation of irreversibly misfolded or unneeded proteins [
30]. Therefore, it serves not only to prevent protein aggregation but also plays a critical role in timely removal of many regulatory proteins, such as those involved in the cell cycle, gene expression, and stress responses [
28]. The major task of the autophagy–lysosomal pathway in proteostasis is the clearance and degradation of deleterious protein aggregates [
31].
Active and quiescent NSCs rely on different pathways to maintain homeostasis in their proteome. Whereas active NSCs display active proteasomes, quiescent NSCs accumulate aggregates in larger lysosomes [
32,
33]. Notably, these differences determine the fate of NSCs. For example, abrogation of lysosomal activity by chemical inhibition or by deletion of the lysosomal master regulator transcription factor EB (TFEB) resulted in activation of quiescent NSCs and delayed return to quiescence in active NSCs, indicating a requirement of lysosomal activity for maintaining NSC quiescence [
33]. Furthermore, active and quiescent NSCs utilize different types of chaperones. Active NSCs express higher amounts of endoplasmic reticulum (ER) unfolded protein response genes responsible for maintaining proteostasis in the ER when newly synthesized protein load exceeds the folding capacity or upon stress-induced accumulation of misfolded proteins [
32]. In contrast, quiescent NSCs exhibit increased expression of T-complex protein 1 (TCP-1) ring complex and its upstream prefoldin complex, which maintain misfolded protein solubility and provide stress resilience in adult NSCs [
32,
34].
Proteostasis capacity declines in aged NSCs, leading to the accumulation of protein aggregates, impaired self-renewal, and lower resilience to stress [
32,
34,
35]. This mainly concerns clearance mechanisms enriched in quiescent NSCs, including a decrease in TCP-1 ring complex and impairments in the autophagy–lysosomal pathway [
32,
34]. The mechanisms responsible for these alterations are not well understood but may involve members of the class O of forkhead box transcription factors (FoxOs), which are transcriptional activators of genes involved in stress-inducible chaperone response, lysosome-autophagy, and the proteasome [
29,
35,
36]. FoxOs participate in nutrient-sensing pathways and activate when the insulin/insulin-like-growth-factor-I (IGF-1) signaling is low, resulting in enhanced autophagy-mediated protein clearance [
29]. Accordingly, deletion of FoxOs in adult NSCs, which impairs autophagy and leads to accumulation of damaged proteins, is associated with hyperproliferation and subsequent depletion of hippocampal NSCs [
35,
37]. This highlights the importance of FoxO-mediated autophagy in restricting the activation of young NSCs to prevent their premature exhaustion [
37]. Conversely, in aged NSCs, autophagy becomes necessary for their activation from quiescence. Here, it has been shown that increasing autophagy, for example by overexpressing TFEB or fasting, alleviates the proliferation deficit of aged NSCs [
32]. Limited knowledge exists on age-related changes in proteasome activity in hippocampal NSCs. A recent study suggests that NSCs in the SVZ preserve proteasome activity throughout their lifespan [
32], in contrast with somatic cells that experience dysfunctional proteasomal function with age [
28]. Overall, available evidence suggests that defective lysosomal degradation plays a major role in NSC aging. Yet, further studies are needed to elucidate the specific mechanisms linking impaired autophagy to NSC dysfunction and their potential implications for preserving their regenerative potential during aging. These include, for example, dysregulation of mitochondria, ROS production, and DNA integrity, which present common points of convergence for autophagy alterations in other stem cell contexts. Considering the dynamic nature of proteostasis and its susceptibility to external factors, interventions such as dietary restriction hold promise in mitigating age-related decline in hippocampal NSCs.
Loss of proteostasis is exacerbated by defective segregation of damaged proteins during asymmetric division of aged NSCs. Similar to other somatic stem cells, NSCs possess mechanisms to prevent the retention of dysfunctional proteins and other harmful cargo during division. This includes a diffusion barrier within the ER membranes that facilitates the segregation of damaged proteins to their differentiating progeny [
38]. However, aging diminishes the efficiency of this barrier so that damaged proteins are passed more evenly, leading to an accumulation of aging factors in aged NSCs with potentially detrimental consequences for their proliferative capacity [
38]. The functionality of this barrier is believed to involve lamins A and B1, components of the nuclear lamina that integrate into the endoplasmic membrane during cell division [
38,
39]. Evidence suggests that decreased expression of lamins and the accumulation of incorrectly processed lamin precursor proteins contribute to barrier weakening with aging [
38,
39,
40]. Furthermore, given their role in stem cell proliferation and fate determination, altered levels of lamins in aging are likely to cause increased quiescence and astrocytic differentiation of aged NSCs [
5,
38,
39,
41,
42]. Yet, lamins have diverse nuclear and cellular functions, ranging from chromatin organization and gene regulation to cytoskeletal organization [
43]. Therefore, further investigation is warranted to establish a causal relationship between their role in barrier strength and the protection of NSCs against aging.
In summary, the dysregulation of proteostasis during aging disrupts the mechanisms that maintain NSC functions and leads to the accumulation of dysfunctional proteins. This can induce cellular stress and inflammatory responses, and impairments in NSC self-renewal and maintenance, which contribute to reduced neurogenesis and compromised plasticity in the aged hippocampus. Understanding and targeting altered proteostasis mechanisms may offer potential strategies to preserve NSC functions and promote healthy hippocampal aging.
2.2. Mitochondria Dysfunction and Metabolic Alterations
Similar to other somatic stem cells, adult NSCs exhibit distinct energy demands depending on their activation state. Several studies suggest that quiescent NSCs derive their energy primarily from glycolysis and fatty acid oxidation (FAO), with little reliance on mitochondrial oxidative phosphorylation (OxPhos), which is the main energy source of active NSCs [
32,
44,
45]. Yet, recent studies showing that pyruvate import into mitochondria and OxPhos are required for the maintenance of NSC quiescence suggest that OxPhos is more important in quiescent NSCs than commonly thought [
46]. In addition, mitochondria of NSCs undergo metabolic and morphological changes during the transition between quiescent and active states [
32,
44,
45]. Recent studies have highlighted the critical role of these changes as active regulators of NSC fate, rather than being passive adaptations to changing energy demands. Perturbation of mitochondrial metabolic rewiring, for example through deletion of YME1L, compromises hippocampal NSC self-renewal and pool maintenance [
47]. Similar outcomes including premature depletion of NSCs, defective neurogenesis, and cognitive impairments were observed upon forced fragmentation of mitochondria by deleting the structural protein MFN1/2 [
48]. This study revealed that mitochondrial dynamics play an upstream regulatory role in NSC fate decisions, with enhanced fusion promoting self-renewal and fragmentation favoring neuronal commitment and differentiation. This is achieved through mitochondrial-to-nuclear retrograde signaling, which triggers a dual gene expression program suppressing self-renewal while promoting differentiation [
48]. It involves a fission-induced increase in oxidative metabolism and elevated ROS, which serves as signaling mechanism for the stabilization of Nrf2—an antioxidant transcription factor—controlling genes involved in neuronal differentiation, redox response, and Notch signaling [
48]. In addition to ROS, mitochondria produce intermediate metabolites which can influence stem cell fate via epigenetic remodeling of nuclear chromatin (see below), including 2-oxoglutarate, acetyl-CoA, and NADH [
49]. 2-oxoglutarate serves as a substrate for DNA and histone methyltransferases, while acetyl-CoA acts as co-factor for lysine acetyltransferases, which reverse the activity of NAD
+-sensing sirtuin-family deacetylases by catalyzing the acetylation of histones and other proteins [
49]. Sirtuins, as metabolic sensors, possess antioxidant and anti-proliferative effects and have been implicated in the maintenance of hippocampal NSCs [
50,
51]. These insights, along with the association of mitochondrial dysfunction with various neurodevelopmental disorders [
52], emphasize the critical role of mitochondria in preserving the functional capacity of adult NSCs.
With advancing age, mitochondria become increasingly dysfunctional, characterized by an accumulation of mitochondrial DNA (mtDNA) mutations, impaired dynamics and respiratory function, and elevated production of ROS, all of which are interrelated and potentially contribute to impaired NSC homeostasis [
50,
53,
54,
55,
56]. It is conceivable that the excessive generation of ROS, coupled with diminished antioxidant capacities in aged NSCs, contributes to the depletion of NSCs observed during aging, as demonstrated in superoxide dismutase-deficient mice or hematopoietic stem cells [
57,
58]. Moreover, suprathreshold accumulation of mtDNA damage and concomitant activation of sirtuins via an elevated NAD
+/NADH ratio may represent a mechanism underlying the astrogliogenic disposal of NSCs in the aged DG [
5,
50].
Recent studies point to a crucial role of lipid metabolism in the regulation of NSCs, although its impact during aging remains to be investigated. In the young SGZ, build-up of lipids via fatty acid synthase-dependent de novo lipogenesis is necessary for the activation of quiescent NSCs and NSC proliferation [
59], while their degradation by FAO is required for maintaining the pool of quiescent NSCs [
60,
61]. Lipids are molecules with manifold functions, ranging from energy storage, membrane assembly, and intra- and inter-cellular communication to gene expression and epigenetic regulation [
62]. Therefore, imbalances in lipid metabolism may contribute to NSC aging in many ways. Since FAO operates in the mitochondrial matrix, it is likely to be affected by age-related mitochondrial dysfunction. This may contribute to perturbed proliferation of hippocampal NSCs and eventually depletion of the NSC pool, as shown after inhibition of FAO in young NSCs [
60]. Likewise, age-related impairments in cellular waste disposal mechanisms such as autophagy may lead to shifts in the lipid composition of aged NSCs. Future studies need to clarify how these metabolic pathways and intracellular lipid composition change with age, what specific metabolites affect NSC functions, and which changes in lipid homeostasis may contribute to age-related decline in the NSC population.
Nutrient-sensing pathways, such as the insulin/IGF-1, adenosine monophosphate-activated protein kinase, and NAD
+-dependent sirtuin pathways, play critical roles in the regulation of adult NSCs and their aging. This involves the previously described FoxO transcription factors acting downstream of the insulin/IGF-1 pathway, which regulate various detoxification processes as a function of nutrient availability and cellular redox state [
63]. Excess nutrients and loss of FoxO promote the activation and accelerate the depletion of hippocampal NSCs [
37], supporting the idea that downregulation of the insulin/IGF-1 pathway is beneficial for long-term maintenance of quiescent NSCs. This is corroborated by studies showing that suppression of insulin/IGF-1 signaling or dietary restriction delay the age-related decline in NSCs [
64,
65,
66,
67]. Moreover, deletion of PTEN, an inhibitor upstream of insulin/IGF-1 signaling that triggers the activation of FoxO3, results in the depletion of the hippocampal NSC pool by increasing proliferation and terminal astrocytic differentiation [
4,
68].
Sirtuins are metabolic sensors activated by NAD
+ during cellular energy depletion. Sirt1 has been implicated in NSC homeostasis, acting as epigenetic repressor of the Hes1 promoter, a key effector of Notch signaling [
67]. Conditional deletion of sirtuin 1 leads to proliferation and rapid exhaustion of NSCs, suggesting a role in maintaining NSC quiescence [
51]. Others have implicated sirtuin 1 in the neuron/astrocyte fate choice during differentiation of embryonic and adult NSCs [
51,
69]. Thus, sirtuins appear to take center stage in metabolic control of adult NSC homeostasis, acting as a nutrient-responsive regulator of NSC self-renewal and differentiation. Although this needs to be confirmed experimentally, it seems highly plausible that the age-related depletion of the NSC pool is driven by the concomitant decline in NAD
+ and sirtuin concentrations [
70].
2.3. DNA Damage and Epigenetic Alterations
To sustain cell-specific gene expression and NSC homeostasis throughout life, it is essential to maintain the stability, integrity, and tightly regulated expression of DNA. A growing number of studies show that DNA is continuously damaged during aging, which, together with the reduced capacity of repair mechanisms, contributes to the progressive decline in stem cell potential [
71]. In addition, aged NSCs experience dysregulation of epigenetic patterns that are critical for maintaining their stem cell features and fate, including changes in DNA methylation, histone modifications, and chromatin remodeling [
72,
73]. In this section, we briefly summarize the available evidence linking DNA damage and epigenetic drift to the functional decline of aged hippocampal NSCs, and refer to other reviews for a more detailed discussion of their possible contributions to stem cell aging [
53,
74,
75,
76,
77].
DNA damage is one of the hallmarks of organismal aging and a leading cause of functional decline in stem cells. DNA damage in NSCs can result from genotoxic stressors of external and internal origin, including toxic metabolites and ROS, and due to their capacity for self-renewal, replication errors, and telomere attrition [
53,
77]. To counteract the accumulation of mutations, cells have a complex network of cellular processes that are activated in response to such damage, collectively known as the DNA damage response. These include DNA repair, cell cycle arrest, or, if the damage is too severe for repair, apoptosis, all aimed at maintaining genomic integrity and preventing the transmission of damaged DNA to progeny. When attempts to repair DNA lesions fail and cells become senescent, they acquire an inflammatory phenotype including the secretion of pro-inflammatory cytokines [
53,
78]. This may not only be detrimental to the damaged NSCs itself, but may also spread to surrounding cells and cause chronic inflammation in the niche, which could contribute to premature aging of neighboring yet unscathed NSCs and their progeny [
40,
53].
In vitro studies have shown that NSCs derived from aged brains carry multiple genomic alterations in the form of deletions or loss of heterozygosity [
79]. Others have observed substantial changes in DNA repair pathways indicative for genotoxic stress which already appear in early stages of hippocampal NSC aging [
8]. Limited research has explored the implications of DNA damage in NSCs, with most of the available data derived from SVZ-NSCs. There, irradiation-induced DNA damage results in a typical DNA damage response, including inflammatory cytokine secretion, proliferative arrest, and premature differentiation, which, however, recovers in the long-term through activation of quiescent NSCs [
80,
81,
82]. Hippocampal NSCs are likely to react similarly to DNA damage, since irradiation of the adult DG results in a severe but reversible impairment of neurogenesis [
83]. The reversibility of the effect suggests that NSCs are differently vulnerable depending on their state, with quiescent NSCs being better protected from genotoxic insults than their cycling counterparts, similar to what has been observed in hematopoietic stem cells [
84]. This idea is further supported by studies in the early postnatal DG, which harbors highly proliferative developmental NSCs that become defective and subsequently deplete upon radiation-induced DNA damage [
85]. In line with a higher exposure of dividing NSCs to genotoxic stressors, recent single-cell transcriptome analyses revealed an increased expression of DNA damage response genes in active versus quiescent NSCs in the SVZ, which is independent of age [
86].
Damage also occurs in mtDNA, even at a much higher rate than in the nucleus [
87]. As mtDNA encodes 13 core OxPhos polypeptides, energy metabolism and respiratory capacity are severely impaired when mtDNA mutations accumulate, which is particularly important for high energy-demanding cells such as active NSCs [
54]. Additionally, suprathreshold accumulation of mtDNA mutations leads to excessive production of ROS, which furthers damage to nuclear and mtDNA, reinforces inflammation, and impacts NSC fate decisions and self-renewal [
50,
53,
76]. Wang et al. [
50] showed that mice lacking the mtDNA repair protein 8-oxoguanine DNA glycosylase exhibit a shift in their differentiation pattern towards the astrocytic lineage due to the spontaneous accumulation of mtDNA damage. This not only highlights the importance of repair mechanisms to preserve critical mitochondrial functions but also provides a mechanism for the aging-related disposal and astrocytic differentiation of hippocampal NSCs [
5].
In addition to preserving the genetic code itself, tight regulation of the epigenetic landscape is critical for maintaining NSC identity and switching between different states and fates in response to environmental signals. This occurs on multiple interconnected layers, including DNA methylation-induced gene silencing, control of gene accessibility by histone modifications and pioneer transcription factors, and the spatial organization of chromatin and nuclear architecture [
88]. According to recent findings, erosion of the cell type-specific epigenome may actually be one of the causes of NSC aging [
73,
75]. As recently shown, aged hippocampal NSCs display a decreased expression of histone lysine demethylases [
8], enzymes that modify the accessibility of regulatory gene regions by removing methylation marks from histone tails. Although this requires further investigation, the resulting accumulation of abnormal histone methylation may lead to dysregulation of gene expression programs essential for homeostasis and self-renewal of NSCs, and thus be a cause of increased NSC quiescence in old age. Others observed tremendous age-related DNA methylation changes in the DG including hypomethylation of CpGs and hypermethylation at CpHs [
89]. Many of these, including those at neurogenesis-associated gene loci, could be reversed by environmental enrichment, an intervention that stimulates adult neurogenesis, illustrating the close relationship between extrinsic factors, epigenetic regulation, and NSC plasticity [
89]. Recently, Tet2, which catalyzes the conversion of 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), was discovered as a possible epigenetic regulator of NSC aging [
90]. Both Tet2 expression and 5hmC levels were observed to decrease in the aged DG. The neurogenic deficit of old mice could be mimicked by downregulating Tet2 in young animals, whereas reinstatement of Tet2 levels by overexpression or heterochronic parabiosis replenished the NSC pool and promoted neurogenesis in the aged DG. Methylated CpGs are bound by other epigenetic modifiers, such as the methyl-CpG-binding domain protein 1 (MBD1), which is expressed by NSCs [
91]. This binding triggers methylation of histone H3, resulting in transcriptional repression of several target genes including that encoding fibroblast growth factor 2 (FGF-2) [
92]. MBD1 deficiency was found to increase proliferation and reduce neuronal differentiation of hippocampal NSCs, accompanied by a repression of lineage differentiation genes and up-regulation of astrocytic genes [
91]. Taken together, these data demonstrate the importance of homeostasis in the epigenetic landscape for maintaining NSC identity and shape during aging and raise the possibility of rejuvenating neurogenic capacity by resetting the epigenome through environmental or pharmacological interventions. Of note, chromatin regions associated with metabolic and transcriptional functions bound by key transcription factors promoting quiescence lose accessibility in aged NSCs, suggesting a novel mechanism of age-related NSC dysfunction [
73].
Pioneer transcription factors are key regulators of cell fate transitions due to their unique ability to access closed chromatin, and thereby initiate reprogramming of silent genes [
93,
94]. This allows other regulatory proteins, including transcription factors, chromatin, and histone modifiers, to access chromatin and assemble into activating or repressive regulatory complexes [
94]. One such pioneering factor is Ascl1, which is required for activation of quiescent NSCs and establishes an open and permissive chromatin state at genes involved in neuronal differentiation [
10,
95,
96,
97]. When Ascl1 was depleted in adult hippocampal NSCs, their behavior resembled that of aged NSCs, which exhibit increased quiescence and decreased propensity to activate [
11]. This implies that decreasing levels of Ascl1, as observed in aged NSCs, drive age-associated changes in NSC dynamics and force them into deeper quiescence. However, the extent to which this involves Ascl1’s pioneer activity warrants further investigation. Sox2, a transcription factor involved in self-renewal and maintenance of developing and adult NSCs, also has pioneer activities [
98,
99,
100]. A recent report suggests a critical role for Sox2 in establishing long-range chromatin interactions in NSCs that control the activity of key cell proliferation genes [
99]. Others proposed a role of Sox2 in establishing a permissive chromatin state at methylated regulatory regions of neurogenic genes in hippocampal stem and progenitor cells (NSPCs), enabling the activation of neuronal differentiation programs once differentiation cues are received [
101]. Moreover, Sox2 cooperates with nuclear structural proteins such as nucleoporins to control the transcriptional landscape of adult NSPCs [
102]. Finally, Sox2 has been shown to interact with histone acetylases to promote proliferation [
100]. Taken together, this evidence establishes Sox2 as a critical player in self-renewal and fate control of NSCs. Given these functions and the fact that Sox2 is downregulated in the aged DG [
25,
103], it is reasonable to assume that aberrant Sox2 function contributes to NSC aging.
In addition to their role as a barrier to the segregation of toxic molecules during NSC division, components of the nuclear envelope such as lamins help to anchor chromatin and control gene expression through interaction with epigenetic modifiers [
104,
105,
106]. During aging, lamin B1 expression decreases in hippocampal NSCs, resulting in a reshaping of the nuclear architecture [
39,
40,
107]. Conditional deletion of lamin B1 in NSCs impairs their proliferation and induces their differentiation, which is accompanied by an upregulation of quiescence genes such as Bmp4 and Id4, indicating repression of these genes by lamin B1 [
39,
42]. Restoration of lamin B1 levels in the aged DG promotes NSC proliferation and neurogenesis [
42]. Yet, it remains to be determined whether these observations are due to the role of lamins as epigenetic regulators of stem cell programs or to the accumulation of deleterious molecules due to a dysfunctional barrier. The fact that lamin B1 deletion leads to detachment of lamin-associated chromatin from the nuclear envelope, redistribution of chromatin, and increased activation of histone marks [
39,
106] suggests that NSC dysfunction in aging is due, at least in part, to lamin B1-associated epigenetic mechanisms.
Overall, the accumulation of DNA damage and epigenetic changes in hippocampal NSCs lead to multiple challenges, such as DNA repair damage, activation of senescence pathways, inflammation, dysregulated gene expression, and thus are primary drivers of NSC aging. Understanding these damage mechanisms in NSCs may offer promising targets for interventions to promote healthy aging in the hippocampus and preserve cognitive functions, especially because loss of epigenetic information is a reversible cause of aging [
108].
2.4. Cellular Senescence
Cellular senescence represents a state of irreversible cell cycle arrest and an endpoint of the aging-related molecular mechanisms discussed before [
109]. It functions as a homeostatic mechanism that inhibits the replication of damaged cells, thereby preventing further expansion of damaged cells [
109]. However, the accumulation of senescent cells has detrimental effects when it comes to aging. As a result of successive cell-intrinsic alterations, senescent cells undergo shifts in their secretome towards a more inflammatory state known as senescence-associated secretory phenotype (SASP) [
53,
77,
110]. The secretion of SASP factors during aging contributes to damage in the surrounding cells, creating a more hostile environment and aggravating inflammation, ultimately resulting in a decline in cellular and tissue functionality [
109].
Senescence is characterized by specific molecular alterations that can be utilized as markers for identifying senescent cells. These comprise heightened β-galactosidase activity, increased expression of cyclin-dependent kinase inhibitors, reduced levels of lamin B1, and the accumulation of DNA damage [
111,
112,
113]. Importantly, these very characteristics are also observed in aged hippocampal NSCs, demonstrating the increasing senescence of these cells with age [
40,
114,
115,
116]. Moreover, at least a few of these alterations appear to be causally linked to NSC dysfunction in aging, such as the upregulation of the cyclin-dependent kinase inhibitor p16INK4a [
116,
117]. However, experimental manipulation of p16INK4a levels in young and aged NSCs revealed that p16INK4a, instead of inducing senescence, prevents aged quiescent hippocampal NSC from being activated by neurogenic stimuli, implying a role in NSC pool maintenance [
116]. On the other hand, a causal relationship between the upregulation of p19ARF and NSC senescence has been demonstrated, albeit only in the SVZ and in a highly artificial senescence accelerated mouse-prone 8 model [
118]. The same study proposed alterations in epigenetic regulation as mechanism underlying the accumulation of p19ARF in aged NSCs, specifically the dysregulation of histone deacetylation [
118].
Epigenetic changes are increasingly recognized as hallmark of senescent cells among the various cell-intrinsic mechanisms that contribute to cellular senescence [
109,
119]. One of them is the previously discussed reduction in lamin B1 [
40,
43,
120], which has been linked to a state of deeper quiescence and transformation of aged NSCs into astrocytes [
5,
39,
41,
42]. Another senescence-associated epigenetic modifier is the PIWI-like RNA-mediated gene silencing 2 (Piwil2), an endoribonuclease crucial for the biogenesis and function of PIWI-interacting RNAs [
121]. Suppression of Piwil2 in NSPCs derived from the postnatal DG induces senescence and astrocytic differentiation, mimicking the situation in aged NSCs whose Piwil2 levels are strongly depleted [
121]. Others revealed an epigenetic Plagl2-Ascl1 signaling pathway at the core of hippocampal NSC activation, whose disruption participates in NSC senescence [
122]. They showed that the loss of Plagl2 in aged NSCs contributes to reduced accessibility of Ascl1 chromatin and, thus, impairs their self-renewal capacity [
10,
11,
95,
96,
97,
122]. Intriguingly, this study revealed that a combination of Plagl2 induction and inhibition of the pro-aging protein Dyrk1a is sufficient to permanently rejuvenate senescent hippocampal NSCs that are otherwise resistant to manipulation of either of these proteins alone or other senescence markers such as p19ARF [
122]. Further research is needed to fully comprehend the implication of these mechanisms in cellular senescence and the aging process.
Altogether, evidence suggests a direct contribution of senescence to the aging-associated impairments of NSCs. The complex interplay between various hallmarks of NSC aging, including the connection between epigenetic alterations and cellular senescence, presents intriguing research areas for further investigation. Furthermore, there is a significant gap regarding the specific molecular mechanisms underlying NSC senescence in the aged DG, underscoring the need for dedicated research to this area. Addressing these gaps is crucial for identifying interventions that can effectively rejuvenate aged NSCs and mitigate the decline in hippocampal neurogenesis and cognition [
123].