Cancer remains a global health challenge, accounting for some 10 million deaths per year worldwide. Despite substantial advancements in prevention and treatment, cancer remains one of the most common causes of mortality, particularly in older adults [
1]. Head and neck cancer (HNC) is a common malignancy that typically presents as a squamous cell carcinoma (HNSCC) originating in the upper aerodigestive tract [
2], with over a half million new diagnoses each year worldwide [
1]. HNC risk factors vary by site and may encompass tobacco and alcohol use, human papillomavirus (HPV) infection and occupational and environmental hazards [
3]. Treatment options include surgery, radiation therapy, chemotherapy, immunotherapy and targeted therapies. Typically, surgery and/or radiation are combined with systemic therapies, depending on the stage and characteristics of the cancer [
4]. As with many cancers, HNC diagnosis at an early stage and appropriate treatment selection are associated with complete response and high overall survival, though significant acute and long-term toxicities affect quality of life [
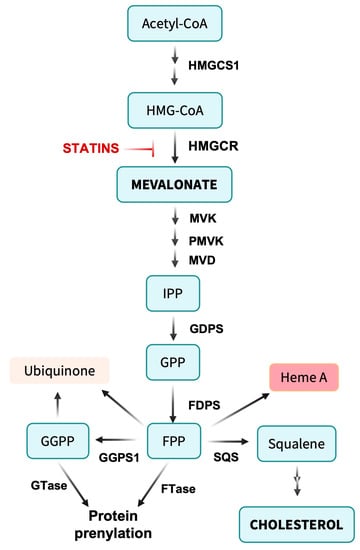
5]. Nonetheless, many patients are diagnosed at an advanced stage and/or recur after their initial therapy, the majority of whom will eventually succumb to their disease. There is a critical, unmet need for more effective primary therapies that can be integrated into current treatment regimens without increasing adverse effects. Competitive inhibitors of the mevalonate pathway enzyme 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, commonly known as statins, may represent an opportunity to both enhance treatment efficacy and lower toxicity [
6,
7,
8].
Most discussion of cholesterol focuses on the deleterious effects of excess biosynthesis and/or dietary uptake such as atherosclerosis [
17]. Nonetheless, cholesterol is a critical component of cell membranes, representing about half of the lipid in plasma membranes, where its content is closely regulated to maintain membrane fluidity and other essential properties in both normal and malignant cells [
18]. Further, in cholesterol-rich membranes, cholesterol may partition into rafts and form complexes with specific proteins [
19]. As such, decreased cholesterol levels upon inhibition of HMG-CoA reductase by statins would be expected to compromise basic cellular functions. However, where cholesterol starvation differentially impacts cancer cells, this may provide a route to increasing the therapeutic index of cancer therapy.
2. Membrane Rafts and Signal Transduction
Given the essential roles for cholesterol in maintaining tumor cell membrane functionality, strategies aimed at inhibiting cholesterol biosynthesis could restrict tumor growth and metastasis [
20]. As an example, the caveolae protein caveolin-1 (CAV1) has been found to modulate the metastatic and invasive capabilities of oral squamous cell carcinoma (OSCC) cells [
21]. Elevated CAV1 expression in metastatic lymph nodes correlates with poor OSCC prognosis. The link to mevalonate pathway metabolism and statins may come via a critical role for cholesterol in lipid raft-dependent functions in cancer cells and tumors [
22]. Lipid rafts function as hubs for signaling proteins, selectively and dynamically regulating their recruitment or exclusion in response to intracellular and extracellular stimuli [
23]. Via concentrating raft-associated proteins and maintaining stable complexes, lipid rafts facilitate signal transduction, a function mediated in part by CAV1. By disrupting rafts and caveolae, statins can have indirect effects on CAV1 and other proteins, leading to CAV1 degradation and altered cell signaling. Of particular significance, lipid raft integrity modulates survival and cell death pathways [
24], suggesting a relationship between lipid rafts and therapy resistance [
25]. Indeed, multiple cancer cell survival and proliferation-related signaling pathways have been linked with lipid rafts [
26]. Providing a potential mechanism for the beneficial effects of statins, disrupting lipid rafts results in the inhibition of the PIK3/Akt signaling pathway, leading to radiosensitization in HNSCC [
27]. A complementary effect may be to limit Akt-induced PD-L1 expression, allowing statins to potentiate anti-tumor immune response [
28].
3. Cholesterol’s Influence on Cancer Cell Proliferation, Survival and Therapy Resistance
Cholesterol’s role in lipid rafts and caveolae in the plasma membrane may impact cancer cell proliferation and survival via effects on signaling by receptors such as HER2 [
29], EGFR [
30] and CXCR4 [
31], as well as transducers and effectors such as PI3K [
32], SRC family kinases [
25] and NOX [
33], along with other regulators [
34]. However, maintaining cholesterol at normal levels in other cellular membranes and subdomains impacts a wide range of pathways important to cancer cell growth, proliferation and resistance. Studies of cholesterol depletion by statins or cyclodextrins suggest that cholesterol helps maintain secretory pathway function, regulates autophagy and supports mitochondrial oxidative phosphorylation [
17,
35,
36]. Other connections appear more indirect. ATAD3A, a protein associated with a wide range of physiological and pathological responses, plays a role in cholesterol metabolism [
37]. Elevated ATAD3A expression has been observed in various cancers, including HNSCC [
38,
39,
40]. In HNSCC, ATAD3A operates as a mitochondrial oncoprotein that stimulates disease progression via the activation of mitochondrial ERK1/2 [
41,
42,
43]. Notably, the ATAD3A-ERK1/2 signaling pathway links to voltage-dependent anion channel 1 (VDAC1) [
41]. VDAC1 promotes the transport of ERK1/2 to the mitochondria, vital for the formation of the ATAD3A-ERK1/2 protein complex in HNSCC cells. Thus, multiple mechanisms may link cholesterol levels to EGFR/PI3K/Akt/mTOR signaling in HNSCC and other cancers [
44,
45,
46].
The calcium-activated chloride channel TMEM16A, previously ANO1, is upregulated in diverse cancers [
47] and commonly overexpressed in HNSCC, which is associated with poor outcomes [
48,
49], establishing it as a therapeutic target. Along with binding to EGFR that may impact HNC proliferation, survival and expression of PD-L1 and thus immune evasion [
47,
50], TMEM16A has been implicated in resistance to conventional therapy and EGFR-targeted agents. TMEM16A upregulation can also suppress apoptosis and promote cisplatin resistance [
51]. Simvastatin impairs TMEM16A channel function—potentially due to cholesterol depletion, though mevalonate-independent mechanisms may be involved—and reduces OSCC cell proliferation in a TMEM16A-dependent manner [
49], suggesting statins as an alternative to TMEM16A inhibitors.
4. Inflammation and Immune Response Modulation
The FDA has approved the anti-PD-1 immune checkpoint blockade (ICB) antibodies nivolumab and pembrolizumab for cisplatin-resistant, relapsed or metastatic HNSCC patients. Clinical trials and research studies have confirmed pembrolizumab’s efficacy and safety, both as a standalone treatment and combined with chemotherapy for recurrent or metastatic HNSCC [
52,
53]. For patients with PD-L1-positive, relapsed, or metastatic HNSCC, pembrolizumab monotherapy is recommended as a first-line treatment [
54]. Despite its potential benefits, ICB therapy faces multiple barriers including intrinsic and acquired resistance and high rates of immune-related adverse events (irAEs). There is considerable interest in combination therapies.
Statins have long been appreciated for their ability to reduce inflammation [
55], and part of this effect may be linked to reducing cellular cholesterol levels. Much like its effects on cancer cells, cholesterol depletion may disrupt rafts and caveolae in immune cells, dispersing receptors and transducers that mediate inflammatory signaling in innate and adaptive immune cells [
56,
57]. As a consequence, a concern would be that lowering cholesterol with statins might reinforce immunosuppression in the tumor microenvironment. Nonetheless, an emerging theme from recent preclinical and patient studies in multiple cancers is that statins potentiate anti-tumor immune responses and/or immunotherapy (e.g., [
58,
59,
60,
61]). Multiple cholesterol-dependent mechanisms may be involved.
Nucleic acid detection in the cytoplasm, which activates the innate immune system, occurs through pattern recognition receptors (PRRs). One such PRR is the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway [
62]. Activation of cGAS by cytosolic DNA induces STING to phosphorylate and activate TBK1 and drive Type I interferon (IFN) pathway activation, which has the potential to induce an effective anti-tumor immune response [
63]. As such, STING agonists are currently being evaluated in multiple contexts as cancer therapeutics [
64]. Along with other effects, Type I IFN signaling may limit cholesterol synthesis, which may then further activate STING via depletion of the ER membrane cholesterol pool [
65]. Simply disturbing cholesterol metabolism as with statins might be sufficient to induce this positive feedback loop.
Other benefits of statins may be to reduce the suppressive influence of excess cholesterol on immune function. Class II major histocompatibility complex (MHC II) molecules are raft-associated proteins expressed on antigen presenting cells such as dendritic cells (DCs) and serve a key role in presenting processed tumor antigen peptides to effector cells, thereby eliciting anti-tumor responses [
66]. Tumor cells can downregulate DC functionality by raising cholesterol levels [
67]. Oxysterol secretion by tumor cells impairs DC migration to lymph nodes and reduces T cell priming [
68].
Cholesterol may also contribute to T cell dysfunction directly via immune checkpoint activation and CD8
+ T cell exhaustion. Membrane cholesterol serves a direct role in stabilizing PD-L1 through its interaction with cholesterol-binding CRAC motifs [
69]. This suggests that reducing cholesterol might be sufficient to interrupt immune checkpoint signaling and/or potentiate immune checkpoint blockade immunotherapy. High cholesterol exposure in the tumor microenvironment is also associated with elevated PD-1 expression by infiltrating CD8
+ T cells [
70] and CD8
+ T cell exhaustion [
71].
Multiple indirect effects of statins can be ascribed to the decreased cholesterol biosynthesis. Intracellular cholesterol depletion may promote cleavage and nuclear localization of sterol regulatory element (SRE)-binding proteins (SREBPs [
72]) that bind SREs, leading to compensatory expression of sterol pathway and coregulated genes. Along with inducing expression of HMGCR and other mevalonate pathway enzymes, SREBPs increase the expression of enzymes for cholesterol biosynthesis (via SREBP-2) and fatty acid and triglyceride biosynthesis (via SREBP-1) as well as the LDL receptor and diverse other proteins linked to lipid metabolism and transport. Insofar as SREBPs are cancer targets [
73,
74], this effect of statins may well be counterproductive beyond simply restoring mevalonate pathway activity. Along these lines, one of the SREBP-dependent proteins induced by statins is the LDL receptor negative regulatory factor PCSK9 [
75,
76]. PCSK9 has emerged as an alternate target for lipid lowering therapy, insofar as inhibiting PCSK9 with antibodies (alirocumab, evolocumab) or siRNA (inclisiran) leads to increased LDLR recycling rather than degradation and greater liver uptake of LDL, lowering circulating cholesterol [
77]. Significantly, PCSK9 has a similar effect on MHC I, leading to its lysosomal transport and downregulation [
78]. Targeting PCSK9 increased tumor cell MHC I expression, promoted CD8
+ T cell tumor infiltration and cytotoxicity and potentiated the effects of PD-1/PD-L1 checkpoint blockade. Effects on CD8
+ T cells may also be direct, as blocking PCSK9 can enhance T cell receptor (TCR) signaling via stabilizing LDLR [
79]. While arguing for targeting PCSK9 in cancer immunotherapy [
80], these considerations also raise the concern that statins have the potential to interfere with immune checkpoint blockade via activation of SREBP and upregulation of PCSK9.