Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Hepatocellular carcinoma (HCC) is one of the most common malignancies in both transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT). The mechanisms of iron-overloading-associated HCC development include the increased reactive oxygen species (ROS), inflammation cytokines, dysregulated hepcidin, and ferroportin metabolism.
- thalassemia
- iron
- transfusion-dependent thalassemia
- Hepatocellular carcinoma (HCC)
1. Iron and Inflammation
When intrahepatic iron overflows, hepatocytes are also stimulated to differentiate into a proinflammatory state through excessive intracellular ferritin by enhancing the NF-κB expression pathway [47]. Prolonged activation of hepatocytes means they can differentiate into myofibroblasts and lead to neovascularization and extracellular matrix formation. Excessive iron molecules are not only deposited in but also injure hepatocytes. They also affect immune system function. Liver, as the largest reticuloendothelial system in the body, contains the largest population of macrophages (Kupffer cells). Macrophages, as scavengers in the body, phagocytize not only external harmful agents, such as bacteria, but also old dysfunctional cells, such as senescent RBCs and destroyed hepatocytes. Thus, they are prone to contain excessive amounts of iron, especially in patients with thalassemia. Excessive amounts of iron induce the macrophage to secrete proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-12 (IL-12) [48]. Together, with the proinflammatory status of hepatocytes, complicated chronic inflammatory interaction ensues. Hepatic stellate cells, endothelial cells, and lymphocytes inside the liver all participate, and cirrhosis begins [49]. Cirrhosis provides an oncogenic environment for HCC formation [50,51,52]. A study of human liver tissue revealed that hepatocytes were polarized to myofibroblasts when the liver iron concentration (LIC) was >60 μmol/g. Cirrhosis ensues when the LIC is >250 μmol/g [53]. Fibrosis develops when the LIC is >400 μmol/g [54].
2. Iron and Reactive Oxygen Species (ROS)
When intracellular iron is excessive, these iron molecules participate in the Fenton reaction (Figure 2) and cause the accumulation of ROS [55]. Healthy cells can detoxify ROS through nonenzymatic and enzymatic antioxidants, such as glutathione, flavonoids, superoxide reductase, catalase, and glutathione peroxidase. If ROS formation exceeds the detoxifying ability of cells, excess ROS can cause genomic instability and reprogram cell metabolism toward carcinogenesis [56]. ROS induce hypoxia-inducible factor-1α expression and related angiogenic gene expression and thus promote angiogenesis [57]. ROS can also promote ERK/NF-kB pathway activation and facilitate tumor cell growth, migration, and deposition of extracellular matrix [58]. To summarize, ROS enhance tumor cell proliferation and survival.
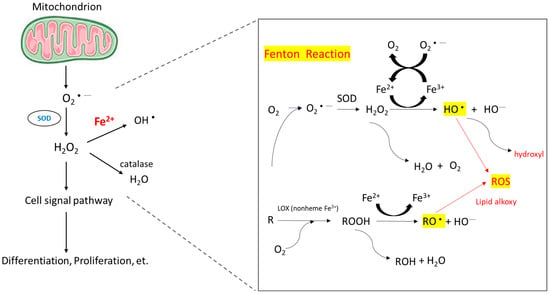
Figure 2. Mechanism of ROS accumulation caused by the Fenton reaction. The reaction of peroxides with Fe2+ to yield soluble hydroxyl (HO•) or lipid alkoxy (RO•) radicals is referred to as the Fenton reaction.
Other than iron, HBV/HCV infection and NASH/NAFLD status provide many oxidative stresses to hepatocytes as well. In NAFLD, elevated mitochondrial fatty acid oxidation and impaired respiratory chain activity make ROS accumulate [59]. It was found that the HBV structural protein-HBx damaged the mitochondrial membrane potential by interacting with the outer membrane and affecting the respiratory complex; ROS production then increased [60]. With chronic HCV infection status, patients usually have a higher liver iron content and thus high ROS production [61]. Thus, in thalassemia patients, co-existence of hepatic viral infection and/or NAFLD with iron overload makes them vulnerable to hepatocarcinogenesis.
3. Hepcidin and Ferriportin
Hepcidin is a polypeptide hormone, isolated from plasma and urine samples with antimicrobial activities by two research groups in 2000 [62,63]. They showed that hepcidin was mainly produced by the liver, followed by the heart, brain, and lung [62]. Almost at the same time, Pigeon et al. tested the hepatic gene expressions in iron-overload mouse models and showed that hepcidin (HEPC), the gene encoding hepcidin, was overexpressed in iron overload hepatic cells. In the report, they also mapped the gene to the human chromosome 19 close to the upstream stimulatory factor-2 (USF2) gene [64]. Lipopolysaccharide, a main component of pathogenic molecules, acts on macrophages, including hepatic Kupffer cells, to induce interleukin-6 (IL-6) production, which in turn induces the overexpression of hepcidin mRNA in hepatocytes [65,66]. In summary, iron overloading, infection, and inflammation can induce hepatocyte hepcidin secretion, which leads to hypoferremia and the anemia of chronic inflammation. Anemia and hypoxia suppress hepcidin expression and result in tissue iron overload. Hepcidin regulates the absorption of dietary iron from the intestine, the release of recycled hemoglobin iron by macrophages, and the movement of stored iron from hepatocytes by blocking the iron transport in the intestinal epithelium, the placenta, and the macrophage [65]. Ferriportin, the only ion exporter in human cells, plays a critical role in hepcidin-regulated iron metabolism, where ferriportin–hepcidin binding leads to the internalization and degradation of ferriportin, and due to the decreased amounts of ferripotin, iron is trapped in hepatocytes, macrophages, and absorptive enterocytes. In consequence, cytoplasmic iron accumulates and reduces the cell uptake of iron. After plasma iron becomes depleted, mainly through hemoglobin synthesis by red cell precursors in the bone marrow, the hepcidin expression is suppressed and restores the iron hemostasis system [67].
As a key regulator of iron metabolism, hepcidin levels are influenced in the pathogenesis of anemia. Inefficient erythropoietic anemias, such as thalassemia, whose clinical manifestation is iron overload anemia, iron deficiency anemia, and congenital erythropoiesis, often have low or abnormal hepcidin relative to the anemia [28]. Kijima et al. examined hepcidin expression in cancerous and noncancerous liver tissues from 40 HCC patients and found that hepcidin mRNA expression was significantly suppressed in cancerous tissues compared to noncancerous liver tissues. It is noncancerous in HCC patients, regardless of the disease state such as tumor differentiation or the time to relapse. There was no significant difference in the mRNA expression of ferripotin and transferrin receptor 2 (TfR2) between cancerous and noncancer tissues [68]. On the other hand, Tan et al. found that hepcidin, TfR2, transferrin (Tf), ceruloplasmin (Cp), and iron regulatory protein 1 (IRP1), isolated in 24 HCC patients with chronic HBV infection, were downregulated compared to the adjacent tumor-free liver tissue and normal liver controls [69]. Similarly, Tseng et al. found that hepcidin, ceruloplasmin, transferrin, and transferrin-phosphorus receptor 2 were downregulated, transferrin receptor 1 was upregulated, and the hepcidin levels consistently correlated with hepatic iron stores in 50 HCC patients [70]. Kessler et al. showed that in cirrhotic tissues, hepcidin expression was lower compared to healthy liver samples in an HBV-related cohort as well as in HCV-infected patients, hepcidin expression was even lower in the HCC samples, and in their mouse models, they showed that hepcidin expression was decreased in early hepatocarcinogenesis as well as in a later stage of murine tumorigenesis [71]. Hepcidin also ameliorates liver fibrosis by inhibiting hepatic stellate cells (HSCs)’ activation, which facilitates liver fibrosis via the degradation of ferroportin in HSCs, increasing Akt phosphorylation and finally prohibiting TGFb1-inducible Smad3 phosphorylation [72].
The underlying mechanisms of suppressed hepcidin in HCC included the suppression of HAMP, TfR2, HJV, ALK2, and/or circular RNA circ_0004913, upregulations of matriptase-2 and/or GDF15, and the inactivation of RUNX3 and/or mutations in TP53 [73]. Hepcidin-induced ferriportin dysregulation has been reported to be associated with an increased risk of cancer, including HCC. FPN overexpression has been reported to be associated with a significant reduction in clonogenic ability, tumor regression, and liver metastasis in breast cancer cells [74,75]. The effects of hepcidin downregulation involve increased cancer proliferation via activation of the CDK1/STAT3 pathway and JAK/STAT pathway in a dependent manner [73,76].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241612654
This entry is offline, you can click here to edit this entry!