Infection with cytomegalovirus (CMV) is highly prevalent in the general population and largely controlled by CD8pos T cells. Intriguingly, anti-CMV T cells accumulate over time to extraordinarily high numbers, are frequently present as tumor-resident ‘bystander’ T cells, and remain functional in cancer patients. Consequently, various strategies for redirecting anti-CMV CD8pos T cells to eliminate cancer cells are currently being developed. This includes conveying the advantageous characteristics of anti-CMV T cells in adoptive cell therapy. Chimeric antigen receptors (CARs) and T cell receptors (TCRs) were transduced into anti-CMV CD8pos T cells to improve the in vivo persistence of CAR/TCR-engineered T cells. Moreover, anti-CMV T cells were activated ex vivo, expanded, and reinfused into glioblastoma patients to directly target CMV peptide epitopes expressed in a subset of glioblastomas.
- CMV
- T cells
- ACT
1. Introduction
2. Strategies Utilizing (Engineered) CMV T Cells in Adoptive Cell Therapy (ACT)
2.1. CMV-CAR T Cells
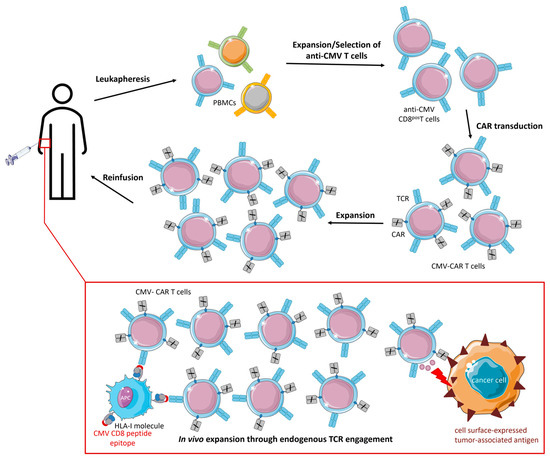
2.2. TCR-Engineered CMV T Cells
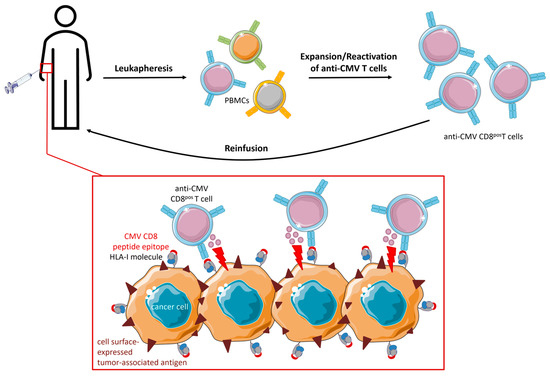
2.3. Direct Targeting of CMV Peptide Epitopes Expressed in Glioblastoma
3. Conclusions
Anti-CMV T cells are of particular interest for cancer (immuno)therapy because they are abundantly present, constantly renewable, and dominate the T-cell pool of the elderly, who are more often affected by cancer. Adoptive cell therapy of engineered CMV T cells aimed to provide optimal co-stimulation and improved in vivo persistence for CAR/TCR-engineered T cells following endogenous TCR interactions with latent virus antigens. Indeed, CMV-CAR T cells demonstrated superior proliferation, survival, and antitumor activity in vivo compared to generic CAR T cells. However, attempts to design TCR-engineered CMV T cells have not been very successful thus far and the competition for surface expression between the endogenous and artificial TCR remains a major challenge. Ex vivo reactivation and ACT of dysfunctional anti-CMV CD8pos T cells in GBM patients proved to be feasible and prolonged PFS and OS.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15153767
References
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034.
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213.
- Dowd, J.B.; Aiello, A.E.; Alley, D.E. Socioeconomic disparities in the seroprevalence of cytomegalovirus infection in the US population: NHANES III. Epidemiology Infect. 2009, 137, 58–65.
- Fowler, K.; Mucha, J.; Neumann, M.; Lewandowski, W.; Kaczanowska, M.; Grys, M.; Schmidt, E.; Natenshon, A.; Talarico, C.; Buck, P.O.; et al. A systematic literature review of the global seroprevalence of cytomegalovirus: Possible implications for treatment, screening, and vaccine development. BMC Public Health 2022, 22, 1659.
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A.H. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals. J. Immunol. 2002, 169, 1984–1992.
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685.
- Komatsu, H.; Sierro, S.; Cuero, A.V.; Klenerman, P. Population analysis of antiviral T cell responses using MHC class I-peptide tetramers. Clin. Exp. Immunol. 2003, 134, 9–12.
- Pita-Lopez, M.L.; Gayoso, I.; DelaRosa, O.; Casado, J.G.; Alonso, C.; Muñoz-Gomariz, E.; Tarazona, R.; Solana, R. Effect of ageing on CMV-specific CD8 T cells from CMV seropositive healthy donors. Immun. Ageing 2009, 6, 11.
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377.
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675.
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. J. Clin. Oncol. 2005, 23, 2346–2357.
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer Res. 2019, 25, 7340–7350.
- Wang, X.; Diamond, D.J.; Forman, S.J.; Nakamura, R. Development of CMV-CD19 bi-specific CAR T cells with post-infusion in vivo boost using an anti-CMV vaccine. Int. J. Hematol. 2021, 114, 544–553.
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of Cellular Immunity against Cytomegalovirus in Recipients of Allogeneic Bone Marrow by Transfer of T-Cell Clones from the Donor. N. Engl. J. Med. 1995, 333, 1038–1044.
- Bollard, C.M.; Kuehnle, I.; Leen, A.; Rooney, C.M.; Heslop, H. Adoptive immunotherapy for posttransplantation viral infections. Biol. Blood Marrow Transplant. 2004, 10, 143–155.
- Leen, A.M.; Christin, A.; Myers, G.D.; Liu, H.; Cruz, C.R.; Hanley, P.J.; Kennedy-Nasser, A.A.; Leung, K.S.; Gee, A.P.; Krance, R.A.; et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood 2009, 114, 4283–4292.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528.
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101.
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973.
- Caruana, I.; Weber, G.; Ballard, B.C.; Wood, M.S.; Savoldo, B.; Dotti, G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin. Cancer Res. 2015, 21, 2952–2962.
- Wang, X.; Wong, C.W.; Urak, R.; Mardiros, A.; Budde, L.E.; Chang, W.C.; Thomas, S.H.; Brown, C.E.; La Rosa, C.; Diamond, D.J.; et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin. Cancer Res. 2015, 21, 2993–3002.
- Wang, X.; Urak, R.; Walter, M.; Guan, M.; Han, T.; Vyas, V.; Chien, S.-H.; Gittins, B.; Clark, M.C.; Mokhtari, S.; et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J. Immunother. Cancer 2022, 10, e003461.
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125.
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus vectored cytomegalovirus vaccine to prevent cytomegalovirus viremia in transplant recipients: A phase 2, randomized clinical trial. Ann. Intern. Med. 2020, 172, 306–316.
- Kalamasz, D.; Long, S.A.; Taniguchi, R.; Buckner, J.H.; Berenson, R.J.; Bonyhadi, M. Optimization of Human T-Cell Expansion Ex Vivo Using Magnetic Beads Conjugated with Anti-CD3 and Anti-CD28 Antibodies. J. Immunother. 2004, 27, 405–418.
- Park, J.H.; Romero, F.A.; Taur, Y.; Sadelain, M.; Brentjens, R.J.; Hohl, T.M.; Seo, S.K. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin. Infect. Dis. 2018, 67, 533–540.
- Hill, J.; Li, D.; Hay, K.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130.
- Tzannou, I.; Papadopoulou, A.; Naik, S.; Leung, K.; Martinez, C.A.; Ramos, C.A.; Carrum, G.; Sasa, G.; Lulla, P.; Watanabe, A.; et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections after Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2017, 35, 3547–3557.
- Heemskerk, M.H.M.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Reprogramming of Virus-specific T Cells into Leukemia-reactive T Cells Using T Cell Receptor Gene Transfer. J. Exp. Med. 2004, 199, 885–894.
- van Loenen, M.M.; Hagedoorn, R.S.; Kester, M.G.; Hoogeboom, M.; Willemze, R.; Falkenburg, J.F.; Heemskerk, M.H. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. 2009, 69, 2034–2041.
- van Balen, P.; Jedema, I.; van Loenen, M.M.; de Boer, R.; van Egmond, H.M.; Hagedoorn, R.S.; Hoogstaten, C.; Veld, S.A.J.; Hageman, L.; van Liempt, P.A.G.; et al. HA-1H T-Cell Receptor Gene Transfer to Redirect Virus-Specific T Cells for Treatment of Hematological Malignancies After Allogeneic Stem Cell Transplantation: A Phase 1 Clinical Study. Front. Immunol. 2020, 11, 1804.
- Peredo-Harvey, I.; Rahbar, A.; Söderberg-Nauclér, C. Presence of the Human Cytomegalovirus in Glioblastomas—A Systematic Review. Cancers 2021, 13, 5051.
- Ahani, N.; Nikravesh, A.; Shirkoohi, R.; Arzenani, M.K.; Rokouei, M.; Eskandani, M.A. Detection of human cytomegalovirus in glioma tumor tissues. Comp. Clin. Pathol. 2013, 23, 1321–1330.
- Stangherlin, L.M.; Castro, F.L.F.; Medeiros, R.S.S.; Guerra, J.M.; Kimura, L.M.; Shirata, N.K.; Nonogaki, S.; dos Santos, C.J.; Silva, M.C.C. Human Cytomegalovirus DNA Quantification and Gene Expression in Gliomas of Different Grades. PLoS ONE 2016, 11, e0159604.
- Crough, T.; Beagley, L.; Smith, C.; Jones, L.; Walker, D.G.; Khanna, R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol. Cell Biol. 2012, 90, 872–880.
- Fornara, O.; Odeberg, J.; Solberg, N.W.; Tammik, C.; Skarman, P.; Peredo, I.; Stragliotto, G.; Rahbar, A.; Söderberg-Nauclér, C. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology 2015, 4, e1036211.
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res. 2014, 74, 3466–3476.
- Sorkhabi, A.D.; Sarkesh, A.; Saeedi, H.; Marofi, F.; Ghaebi, M.; Silvestris, N.; Baradaran, B.; Brunetti, O. The Basis and Advances in Clinical Application of Cytomegalovirus-Specific Cytotoxic T Cell Immunotherapy for Glioblastoma Multiforme. Front. Oncol. 2022, 12, 818447.
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053.
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E.; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256–264.