Multiple Myeloma is a genetically heterogeneous disease, arising and progressing through the appearance and accumulation of a tangle of genomic aberrations. In the last decade, cheap and wide applicable sequencing technologies allowed significant advantages in its biological knowledge. Here we focus on genomic events that drive plasma cell disorders.
- Multiple Myeloma
- genomic aberrations
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Multiple myeloma (MM) is a plasma cell disorder, accounting in term of prevalence for about 10% of hematological malignancies. MM is a genetically heterogenous disease arising from the accumulation of genomic events over the years [1]. The model of clonal progression from a pre-malignant stage of monoclonal gammopathy of uncertain significance (MGUS) through an asymptomatic stage named smoldering multiple myeloma (SMM) to MM and plasma cell leukemia (PCL) is well studied [2–4]. The accumulation of mutations and chromosome abnormalities over the time [5] favor us in considering MM a model of cancer genesis and evolution.
The process of tumor genesis and progression is triggered by several recurrent events at the genome and chromosome level. Some events typically occur at diagnosis, whereas others could be detected in most patients in late stages of the disease [6]. Furthermore, the concurrence of several and genetically heterogeneous subclones within MM cells—in a context of selection pressure—complicates the interpretation of the genetic of the tumor for intrinsic spatial [7,8] and population heterogeneity [9–11]. In recent years, whole-exome (WES) and whole-genome sequencing (WGS) studies highlighted the complex genomic aberrations underlying the pathogenesis of the disease [12–15].
2. Landscape of Genomic Damage in MM
2.1. Timing of Genome Aberrations in MM
MM has been studied by systematic analysis of target sequencing, WES, and single nucleotide polymorphisms (SNP) array, to identify DNA aberrations recurring at high and intermediate frequencies, with the aim to appreciate pathogenetic dysregulated pathways and potential therapy targets. The analyses revealed in MM a somatic mutation rate of 1.6 mutations per MB [11,13,14,16–22].
Plasma cell disorders are genetically heterogeneous diseases and their clonal progression occurs in sequential phases [11]. Hyperdiploid (HD) events are characterized by the acquisition of chromosome trisomy. Frequently, trisomy occurs in chromosome 3, 5, 7, 9, 11, 15, 19, 21 [19,23]. HD is the main event identified in early stage MM [11]. Non-HD events occurring in early stage MM pathogenesis are characterized by the well-known translocations typically affecting the genes encoding immunoglobulin (Ig) heavy chains (IGH), mainly t(4;14), t(6;14), t(11;14), t(14;16), and t(14;20) [19]. HD MM carries a lower prevalence of primary translocations, as opposed to non-HD MM. Due to their different pathogenesis, they are recognized as the two main genetic subtypes of MM. Secondary events, detected generally in later stages, are essential for tumor progression and consist of chromosomal translocations, copy-number variations (CNV) and single-nucleotide somatic variations that affect several signaling pathways, cell-cycle regulators, and DNA-damage repair mechanisms [19].
Aktas Samur recently described the chronology of copy number aberrations in 164 MGUS and 336 newly diagnosed MM. HD MM seems to originate from different events if compared with non-HD MM. In HD MM, CNVs are also clonal at the MGUS stage, ratifying that these are early events leading to transformation of normal plasma cell to pathologic MGUS cell but are not sufficient to generate MM proliferating cells. The proposed model of HD myeloma genesis and progression suggests the initial gain of at least two copy of chromosome 9, 15, and 19 also present in the MGUS stage; some patients then acquires trisomy of chromosome 6, or chromosome 18, or chromosome 21, while other patients lose chromosome 13 and/or gain 1q (about 20% of patients show no additional CNVs). In non-HD MM, IgH translocations are the earliest event, also identified in MGUS, and most patients do not accumulate other additional changes. In both groups, late catastrophic genomic events can occur, leading to complex deletions, irrespective of initial aberrations [24]. A mention must be referred to TP53 aberrations. Both mutations of the TP53 gene and deletions of chromosome 17p13 containing the TP53 gene are late events in MM history [9,25]. Deletion of 17p occurs with a frequency of about 9.5% in newly diagnosed symptomatic MM patients, rising up to 50% in primary plasma cell leukemia and to 75% in secondary plasma cell leukemia patients [19,26]. Similarly, TP53 mutations occur in 5–8% of newly diagnosed symptomatic MM patients, rising up to 25% in PCL [26–30]. TP53 mutations usually appears after or concurrently with deletion 17p13 [31]. Conversely, TP53 mutations are identified in one third of 17p deleted patients [27].
Damages arise in clone or sub-clone in MM. Early mutations in MM evolution are expected to be clonal. Mutations mostly occurring at the sub-clonal level arise later in MM progression. Nevertheless, several driver gene mutations can be found in MM at the sub-clonal level, underlining the heterogeneity of the cell population. For example, Bolli et al. identified sub-clonal mutations of the driver genes KRAS, NRAS, TP53. TP53 was sub-clonal in 26.7% and clonal in 11% of case. No significant differences were identified in prognosis when a mutation was identified at the clonal or sub-clonal level [5]. Data are lacking about clonal selection induced by treatment in MM. An analysis by Corre et al. evaluated 43 homogeneously treated MM patients for mutational profiles at the time of diagnosis and at relaps [32]. No specific or significant events in mutational status were found after treatment neither at relapse. Every patient seems to evolve differently, with chemoresistance and relapse driven by acquired mutations or selection of pre-existing subclones [32].
2.2. Mechanisms of DNA Damage in MM
In MM, genomic aberrations arise from various types of DNA damage (Figure 1). Nucleotide damage (ND) and single strand breaks (SSB) hesitate in single base substitutions. Double strand breaks (DSB) often alter chromosome structure.
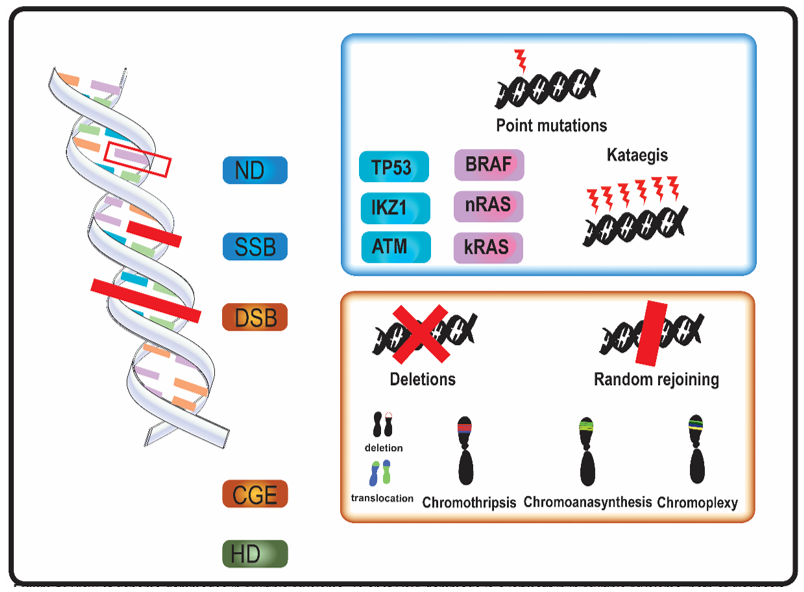
Figure 1. Overview of genome aberrations in Multiple Myeloma (MM). DNA damage is a leitmotif in in multiple myeloma, both at diagnosis and relapse. Nucleotide damages (ND) and single strand breaks (SSB) cause base substitutions that alter proteins and cell function. Frequent point mutations of clinical significance occur in genes involved in DNA integrity (in blue, e.g., TP53) and in cell cycle genes (in purple, e.g., kRAS). A DNA-level complex event named kataegis—a cluster of hundreds to thousands of point mutation in less than few megabases—was occasionally described in multiple myeloma. Double strand breaks (DSB) and complex genome events (CGE) widely alter genome and chromosome structure. Other than classical karyotype events (i.e., sub-chromosome deletions and translocations), in multiple myeloma were described chromothripsis, a one-step catastrophic event in which one or few chromosome were fragmented and randomly rejoined, with casual gain and losses of DNA (blue and red in figure, respectively); chromoanasynthesis a sub-chromosomal local cluster of duplications and triplications on one single chromosome (green clusters in figure); chromoplexy, a series of chained, complex inter- and intra-chromosome translocations, involving 1 to 8 chromosomes, with frequent loss of material at breakpoints. Alterations of ploidy and translocations are the 3rd category of genome abnormalities in MM, and hyperdiploidy (HD) is the main alteration in chromosome number.
Mutational events as kataegis and complex genome events (CGE) were described in MM [11,16–22]. Kataegis and, in general, “apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like” (APOBEC) family-related signatures (a signature with C > T mutations in regions rich in 5-methyl-CpG and another one consisting C > T and C > G mutations in TCN trinucleotide repeats, which results from overactivation of cytidine deaminase activity) were observed in MM [5,11,19].
All the CGE described in MM, chromothripsis, chromoanasynthesis, and chromoplexy—also grouped together under the name of chromoanagenesis—arise from DSB [33]. SNP-microarray and WGS are able to identify these CGE also when localized genomic aberrations are not detected by conventional cytogenetic and Fluorescent in situ hybridization (FISH) analysis [17,34,35]. Genomics events bring a rapid genome variation and could also play a role in genome adaptive evolution [36].
Alterations of ploidy and translocations are frequent as a result of a disfunction in control mechanisms in late mitosis [37]. HD and translocations are the most common events historically described as driver events in MM.
Table 1 reports most frequent DNA damage events, as described in recent genomic studies.
Table 1. List of aberration deriving from genome or DNA damage in multiple myeloma, sorted by incidence or prevalence. Some digits may vary from original studies due to rounding.
|
Aberration |
Incidence at Diagnosis |
Prevalence |
Citations |
|
Single nucleotide variants |
|||
|
MAPK pathway (mostly kRAS, nRAS, BRAF) |
|
40% |
[10,11,38] |
|
NF-KB pathway |
|
20% |
[10,11,38] |
|
DNA-repair pathway |
|
15% |
[38] |
|
TP53 mut |
2–4% |
20–40% |
[27,38–41] |
|
Chromosome-level events |
|||
|
1q gain |
31–37% |
|
[24,42,43] |
|
+15 |
37% |
|
[43] |
|
+9 |
36% |
|
[43] |
|
+19 |
33% |
|
[43] |
|
+5 |
33% |
|
[43] |
|
+3 |
33% |
|
[43] |
|
+11 |
25% |
|
[43] |
|
+7 |
21% |
|
[43] |
|
6p gain |
15% |
|
[42,43] |
|
+21 |
12% |
|
[43] |
|
11q gain |
10% |
|
[42,43] |
|
+X |
9% |
|
[43] |
|
13q loss |
59–60% |
|
[24,43] |
|
16q loss |
28–35% |
|
[42,43] |
|
1p loss |
24–30% |
|
[42,43] |
|
14q loss |
23–39% |
|
[24,42,43] |
|
8p loss |
22–25% |
|
[42,43] |
|
6q loss |
21–33% |
|
[42,43] |
|
−22 |
18% |
|
[43] |
|
16p loss |
15% |
|
[42] |
|
12p loss |
13–15% |
|
[42,43] |
|
−20 |
12% |
|
[43] |
|
del(17p) |
7–10% |
80% |
[40,41,43] |
|
Hyperdyploidy |
|
|
|
|
Hyperdyploidy |
50% |
|
[19,24,44–46] |
|
Translocations |
|
|
|
|
t(11;14)(q13;q32) IGH/CCND1 |
15% |
|
[19] |
|
t(14;16)(q32;q23) IGH/MAF |
5% |
|
[19] |
|
t(6;14)(p21;q32) IGH/CCND3 |
1–2% |
|
[19] |
|
t(14;20)(q32;q12) IGH/MAFB |
1% |
|
[19] |
|
translocations involving MYC |
15–20% |
|
[19,47,48] |
|
Rare and complex events |
|
|
|
|
Kataegis |
3% |
|
[19] |
|
Chromothripsis |
1–24% |
|
[15,18,20] |
|
Chromoanasynthesis |
Anecdotical-19% |
Anecdotical |
[15,16,22] |
|
Chromoplexy |
4–11% |
4% |
[15,17,18] |
References
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk‐stratification and management. J. Hematol. 2020, 95, 548–567, doi:10.1002/ajh.25791.
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispensieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. Engl. J. Med. 2018, 378, 241–249, doi:10.1056/NEJMoa1709974.
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispensieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. Engl. J. Med. 2007, 356, 2582–2589, doi:10.1056/NEJMoa070389.
- van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018, 78, 2449–2456, doi:10.1158/0008-5472.CAN-17-3115.
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.T.; Mudie, L.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018, 32, 2604–2616, doi:10.1038/s41375-018-0037-9.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Rev. Cancer 2012, 12, 335–348, doi:10.1038/nrc3257.
- Coffey, D.G.; Wu, Q.V.; Towlerton, A.M.H.; Ornelas, S.; Morales, A.J.; Xu, Y.; Green, D.J.; Warren, E.H. Ultradeep, targeted sequencing reveals distinct mutations in blood compared to matched bone marrow among patients with multiple myeloma. Blood Cancer J. 2019, 9, 1–4, doi:10.1038/s41408-019-0238-0.
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Commun. 2017, 8, doi:10.1038/s41467-017-00296-y.
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Rev. Clin. Oncol. 2017, 14, 100–113, doi:10.1038/nrclinonc.2016.122.
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Cordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101, doi:10.1016/j.ccr.2013.12.015.
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Commun. 2014, 5, 1–13, doi:10.1038/ncomms3997.
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouelette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93, doi:10.1038/s41586-020-1969-6.
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218, doi:10.1038/nature12213.
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501, doi:10.1038/nature12912.
- Rustad, E.H.; Yellapantula, V.D.; Glodzik, D.; Maclachlan, K.H.: Diamond, B.; Boyle, E.M.; Ashby, C.; Blaney, P.; Gundem, G.; Hultcrantz, M.; et al. Revealing the Impact of Structural Variants in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 258–273, doi:10.1158/2643-3230.bcd-20-0132.
- Berry, N.K.; Dixon-McIver, A.; Scott, R.J.; Rowlings, P.; Enjeti, A.K. Detection of complex genomic signatures associated with risk in plasma cell disorders. Cancer Genet. 2017, 218–219, 1–9, doi:10.1016/j.cancergen.2017.08.004.
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzales, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Commun. 2019, 10, doi:10.1038/s41467-019-11680-1.
- Kaur, G.; Gupta, R.; Mathur, N.; Rani, L.; Kumar, L.; Sharma, A.; Singh, V.; Gupta, A.; Sharma, O.D. Clinical impact of chromothriptic complex chromosomal rearrangements in newly diagnosed multiple myeloma. Res. 2019, 76, 58–64, doi:10.1016/j.leukres.2018.12.005.
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Commun. 2015, 6, doi:10.1038/ncomms7997.
- Magrangeas, F.; Avet-Loiseau, H.; Munshi, N.C.; Minvielle, S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011, 118, 675–678, doi:10.1182/blood-2011-03-344069.
- Lee, K.J.; Lee, K.H.; Yoon, K.A.; Sohn, J.Y.; Lee, E.; Lee, H.; Eom, H.S.; Kong, S.Y. Chromothripsis in Treatment Resistance in Multiple Myeloma. Genomics Inform. 2017, 15, 87–97, doi:10.5808/gi.2017.15.3.87.
- Saxe, D.; Seo, E.J.; Beaulieu Bergeron, M.; Han, J.Y. Recent advances in cytogenetic characterization of multiple myeloma. J. Lab. Hematol. 2019, 41, 5–14, doi:10.1111/ijlh.12882.
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mullingan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007, 67, 2982–2989, doi:10.1158/0008-5472.CAN-06-4046.
- Aktas Samur, A.; Minvielle, S.; Shammas, M.; Fulciniti, M.; Magrangeas, F.; Richardson, P.G.; Moreau, P.; Attal, M.; Anderson, K.C.; Parmigiani, G.; et al. Deciphering the chronology of copy number alterations in Multiple Myeloma. Blood Cancer J. 2019, 9, 1–10, doi:10.1038/s41408-019-0199-3.
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and targeting of TP53 pathway in multiple myeloma. Oncol. 2019, 9, doi:10.3389/fonc.2018.00665.
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-Dependent Activation of a MYC Transgene Induces Multiple Myeloma in a Conditional Mouse Model of Post-Germinal Center Malignancies. Cancer Cell 2008, 13, 167–180, doi:10.1016/j.ccr.2008.01.007.
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976, doi:10.3324/haematol.2010.023697.
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzales, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosom. Cancer 2011, 50, 765–774, doi:10.1002/gcc.20899.
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M,; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597, doi:10.1182/blood-2018-03-840132.
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Bandini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353–21361, doi:10.18632/oncotarget.7241.
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610, doi:10.1038/bcj.2017.76.
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647, doi:10.1038/s41375-018-0153-6.
- Poot, M. Genes, proteins, and biological pathways preventing chromothripsis. Methods Mol. Biol. 2018, 1769, 231–251, doi:10.1007/978-1-4939-7780-2_15.
- Berry, N.K.; Scott, R.J.; Rowlings, P.; Enjeti, A.K. Clinical use of SNP-microarrays for the detection of genome-wide changes in haematological malignancies. Rev. Oncol. Hematol. 2019, 142, 58–67, doi:10.1016/j.critrevonc.2019.07.016.
- Sawyer, J.R. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011, 204, 3–12, doi:10.1016/j.cancergencyto.2010.11.002.
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A piece of the macroevolution scenario. Cytogenet. 2020, 13, 1–9, doi:10.1186/s13039-020-0470-0.
- Onodera, N.; McCabe, N.R.; Rubin, C.M. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia. Blood 1992, 80, 203–208, doi:10.1182/blood.v80.1.203.203.
- Walker, B.A.; Boyle, E.M.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. Clin. Oncol. 2015, 33, 3911–3920, doi:10.1200/JCO.2014.59.1503.
- Petrackova, A.; Minarik, J.; et al. Diagnostic deep-targeted next-generation sequencing assessment of TP53 gene mutations in multiple myeloma from the whole bone marrow. Br. J. Haematol. 2020, 189, e122–e125, doi:10.1111/bjh.16547.
- Fonseca, R.; Blood, E.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575, doi:10.1182/blood-2002-10-3017.
- Tiedemann, R.E.; Gonzalez-Paz, N.; et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008, 22, 1044–1052, doi:10.1038/leu.2008.4.
- Avet-Loiseau, H.; Li, C.; et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 2009, 27, 4585–4590, doi:10.1200/JCO.2008.20.6136.
- Walker, B.A.; Leone, P.E.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, doi:10.1182/blood-2010-04-279596.
- Smadja, N. V.; Fruchart, C.; et al. Chromosomal analysis in multiple myeloma: Cytogenetic evidence of two different diseases. Leukemia 1998, 12, 960–969, doi:10.1038/sj.leu.2401041.
- Debes-Marun, C.S.; Dewald, G.W.; et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia 2003, 17, 427–436, doi:10.1038/sj.leu.2402797.
- Chng, W.J.; Van Wier, S.A.; et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood 2005, 106, 2156–2161, doi:10.1182/blood-2005-02-0761.
- Avet-Loiseau, H.; Gerson, F.; et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086, doi:10.1182/blood.V98.10.3082.
- Walker, B.A.; Wardell, C.P.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191, doi:10.1038/bcj.2014.13
This entry is adapted from the peer-reviewed paper 10.3390/genes11121453