Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Parasitology
Kala-azar, also known as visceral leishmaniasis (VL), is a disease caused by Leishmania infantum and L. donovani. Patients experience symptoms such as fever, weight loss, paleness, and enlarged liver and spleen. The disease also affects immunosuppressed individuals and has an overall mortality rate of up to 10%.
- kala-azar
- visceral leishmaniasis
- Leishmania infantum
- Leishmania donovani
- pathogenesis
1. Infection
Kala-azar, or visceral leishmaniasis (VL), is the second most lethal tropical and subtropical disease and seventh in the loss of disability-adjusted life years [1,2]. It is caused by the protozoa, Leishmania infantum and L. donovani and is transmitted by the bite of infected sand flies. VL caused by L. infantum is a zoonosis involving a variety of mammals, especially the dog, distributed across Central Asia, the Middle East, the Mediterranean Basin, and Central and South America. The disease caused by L. donovani is restricted to humans in South Asia and East Africa [3]. VL has a protracted course of clinical development with fever, weight loss, paleness, and hepatosplenomegaly. Five to ten percent of patients die of this disease, resulting from the lethal complications of bacterial co-infections and hemorrhages. Anemia, neutropenia, thrombocytopenia, hypoalbuminemia, hyperglobulinemia, high C-reactive protein (CRP), and high erythrocyte sedimentation rate (ESR) are regularly noticed. It is a significant opportunistic infection in immunocompromised individuals, particularly those living with HIV or taking immunosuppressive drugs [4]. While transmission has been rapidly and consistently falling in recent years in South Asia [5,6], the incidence in Brazil has a worrisome trend toward a gradual increase in mortality [7].
The development of VL is more exception than rule after exposure to viscerotropic Leishmania. Indeed, a certain proportion of the human population in a given VL endemic area are leishmanin skin tests positive, indicating previous Leishmania infection, but only a small proportion of them have a history of the disease development [8-11]. Based on a population-based leishmanin skin-test study, the ratio of asymptomatic to symptomatic infection was estimated to be more than 200:1 [12].
The early preclinical stages of human infection by viscerotropic Leishmania have defied investigation in situ due to the elusive, long and variable lengths of the incubation period. The early events of infection and host response are available from a large body of extensive work on experimental leishmaniasis in vitro and in animal models. They are included for discussion with the caveat that they may or may not reflect human infection in the real world. Notably, infection of mice or dogs with L. donovani or L. infantum does not reproduce all the clinical symptoms and signs of human visceral leishmaniasis.
In animal models, infected sand flies have been found to inoculate infective promastigotes of various Leishmania spp., e.g., L. infantum, L. donovani, and L. amazonensis into the skin. Most of them are killed in a few minutes by complement-mediated lysis, leaving few survivors phagocytosed by neutrophils or monocytes [13]. The cutaneous species, L. major, encounter the phagocytes via their pathogen recognition receptors (PRR) when successfully infecting C57BL/6 mice [14-16]. It is not known if the infected neutrophils may present antigens directly to T cells, as reported for viral antigens in human neutrophils in vitro [17] or if they are phagocytized by neutrophils that undergo apoptosis and then are ingested by monocytes—the Trojan horse hypothesis [18]. L. major is also directly phagocytized by human monocytes, consistent with the leishmanial intracellular parasitism of mononuclear phagocytes known for successful infection, as demonstrated in humanized transgenic mice [19]. Leishmania antigens are presented by infected macrophages to B- and T- cells, and adaptive immunity develops [20]. The interaction of Leishmania with dendritic cells (DC) has been reviewed [21] in the context of their significance as the antigen-presenting cells in T cell immunity [22]. Leishmania–DC interactions in relation to the lasting immunity, which patients develop after the cure of leishmaniasis is of great interest for further investigation.
The migratory pathway of L. infantum and L. donovani infection is expected to begin from the skin and, via the regional lymph nodes, to arrive at the final destinations of spleen, bone marrow, and liver. A struggle is predicted to emerge between permissive versus protective innate and adaptive immune response, determining host susceptibility or resistance during this Leishmania migration. The battle begins involving the armamentarium, composed of the parasite’s molecules, such as surface lipophosphoglycan and GP63 (zinc metalloprotease), and the host’s molecules initially confronting the parasite: Toll-like receptors (TLRs) (TLR2, TLR4, and TLR9) and other types of receptors, together with the antimicrobial hydrolases/peptides of infected macrophages, reactive oxygen species and nitric oxide produced by these and other mononuclear innate immune cells. Disease ensues when parasite molecules subvert the host response, turning it amenable to Leishmania intracellular survival via several levels of molecular interactions, e.g.: [A] induction of regulatory cytokine IL-10 by parasitized macrophages; [B] inhibition of Th1-acquired immune response and IFN-γ release; and [C] promotion of Th2 T cells and regulatory T cell (Tregs) clonal expansion by further secretion of IL-10, IL-4 and TGF-β regulatory cytokines. This process finally results in T cell anergy and leads to disease development. Infected neutrophils and monocytes/macrophages were observed in the patients’ blood, presumably serving to spread amastigotes to other infection sites and facilitating transmission to other hosts as female sand flies take bloodmeals [23]. The host may win the battle against the infection when macrophages that phagocytose Leishmania produce IL-12, thereby stimulating the expansion of proinflammatory Th1 T cell clones for IFN-γ release, gaining the ability to kill the parasites. After this point, cell-mediated immunity is expected to be firmly established to control parasite proliferation. The host–parasite struggle for viscerotropic Leishmania to establish parasitism in the mammal hosts has been elegantly reviewed by previous workers [24-27].
Disease development is modulated by the parasite–vector–mammalian host interactions. Genomic studies of viscerotropic versus cutanotropic Leishmania delineate the significance of the products of the A2 gene family in parasite visceralization. A mutation in the Ras-like Rag C GTPase in the mTOR pathway of BALB7c mice may contribute to the control of Leishmania visceralization [28]. These observations offer clues for investigation to explain why L. donovani variants cause human cutaneous leishmaniasis instead of VL in Sri Lanka [29]. Finally, whole-genome sequence analyses of 109 L. infantum isolates found parasite sequences that are strongly associated with the phenotypes of disease severity, such as mortality and kidney failure [30]. However, observations on the early preclinical events in human infection will be needed to fully understand the important question of how the disease is initiated. This might eventually be accomplished through the use of controlled human infection models (CHIM), as performed with SARS-CoV-2 [31] notwithstanding the limitations of this approach. The development of more sophisticated human 3D organoid culture systems [32] may be useful to study infection, if vasculature can be introduced for the influx of immune cells necessary for studying infection and immunity. Another useful approach under development is the digital microfluidic sensing to monitor in real time inflammation and over-reactive immune response [33].
Evidence indicates that human hosts contribute to the development of VL. Several studies have demonstrated that the human genetic background plays a role in the fate of the established infection by modulating its development into a disease state [34-37]. There is evidence of age-dependent susceptibility to VL, as indicated by the higher proportion of disease among the youngest in a given population [9,10,38]. Patients at the extreme ends of the age groups also tend to have higher parasite burdens, presumably due to their weaker immunity [39]. Both disease rate of incidence and parasite loads are higher in males at reproductive age than in the other demographic groups [39-43], leading to the proposal that male sex hormones, including testosterone and dihydrotestosterone, may play a role in fueling the infection [44]. Another important host factor influencing the development of disease is the immunity due to prior infection. Solid immunity is clearly generated among those with previous Leishmania infections in an immunocompetent population, regardless of symptomatic or asymptomatic individuals, as both reactivation and reinfection are rarely seen [45,46]. On the same track, acquired immunosuppression strongly influences evolution of the infection in this population from asymptomatic to symptomatic [47]. Finally, malnutrition has long been considered as a risk factor for neglected tropical diseases, although its role in human VL is unclear [9,10,38].
Sand flies can also modulate the outcome of Leishmania infection when delivering infective promastigotes experimentally into the skin of mice with diverse species of Leishmania. Via the bites of sand flies, it has been shown that their saliva is rich in pharmacologically active substances, which modulate vasodilation, anticoagulation and immune response in favor of Leishmania successful infection. Sand fly gut microbiota may also play a certain role [48]. The success or failure of Leishmania infection in mice is ultimately determined by the dose of infective parasites delivered by the vectors [49,50]. Figure 1 shows the likely evolution of VL from the sand fly bite to disease development.
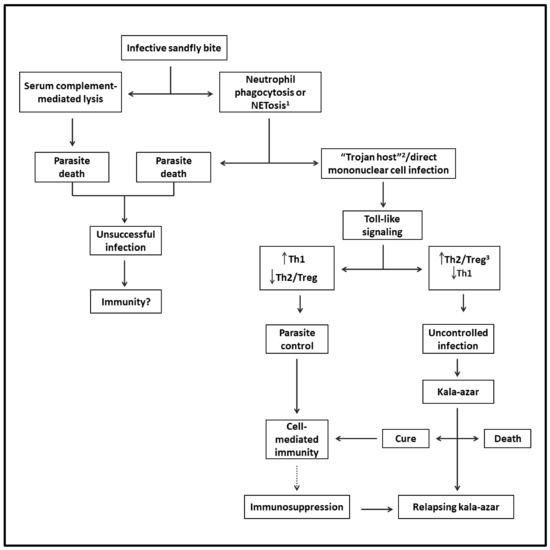
Figure 1. Flowchart depicting the likely evolution of VL from infection to disease: Via the bite of an infected sand fly, infective parasites are released into the skin. Some perish, while viable parasites reach their host cells directly via phagocytosis by mononuclear phagocytes and/or via phagocytosis by neutrophils (Trojan host), followed by engulfment by mononuclear cells. After triggering Toll-like intracellular signaling in either case, the infection is controlled by Th1- and cell-mediated immunity. However, the parasites may overcome this host response to replicate by mediating the generation of Th2 and Treg responses, progressing to VL. The patients may die or be cured, leading to lasting immunity to reinfection. Some patients may suffer from relapsing VL due to suppression of cell-mediated immunity preventing a complete cure. 1 Regulated cell death with the formation of neutrophil extracellular traps (NET). 2 A mechanism of intracellular cell entry through the phagocytosis of neutrophils with engulfed microbe by mononuclear phagocytes. 3 T-regulatory cells.
2. Disease
Successful infection leads to unchecked proliferation of amastigotes in the mononuclear phagocytes, accounting for the disease state of VL. That is marked by systemic innate response with an increase in the plasma level of proinflammatory cytokines and the regulatory cytokine, IL-10 [51,52]. Parasites must overcome host resistance in order to proliferate for the disease to ensue. The emergence of Th2 and Treg susceptible phenotype over Leishmania-specific acquired cellular immunity is expected, as indicated by the failure of immune T cells to proliferate and to produce IFN-γ in clinical cases [53-57]. Since Leishmania produce no toxins to account for the pathology of the disease seen, the innate host immune response is solely responsible for the clinical symptoms observed [58]. Indeed, there is a positive correlation between severity of the disease and parasite burdens [39,59]. Figure 2 shows some examples of children with various signs and symptoms of VL.
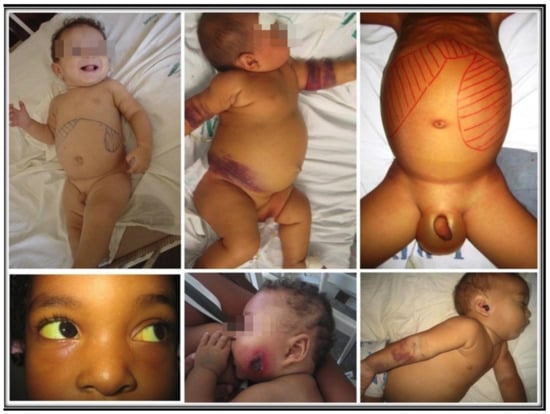
Figure 2. Uncomplicated, complicated, and lethal VL. Top left: an uncomplicated disease with hepatosplenomegaly and paleness. Top center: extensive bruising. Top right: large hepatosplenomegaly, with ascites and edema of the scrotum. Bottom left: scleral jaundice. Bottom center and right: Pseudomonas aeruginosa secondary infection in the face and the ear.
3. Constitutional Symptoms
Table 1 summarizes the hypothetical pathogenesis of VL. VL’s signs and symptoms overlap with those of overproducing proinflammatory cytokines, like IL-6, IL-1β, TNF-α and IL-8 as well as the regulatory cytokines, IL-10 and TGF-β. All these cytokines are produced via signal transduction triggered by Leishmania interaction with PRRs after infection of macrophages [60]. IL-6 is thought to have the broadest spectrum of impact due to the nature of its receptors. IL-6 interacts with both membrane-bound (mIL-6R) and soluble IL-6 receptors and the co-receptor gp130, accounting for its pleiotropic activities [61]. The binding of IL-6 to mIL-6R and gp130 induces the synthesis of the acute phase reactants (APR) in hepatocytes [62] and also the overexpression of tissue factors in endothelial cells and monocytes, triggering the coagulation cascade [63,64,65]. The gp130 co-receptor of IL-6 is expressed on the plasma membrane of most cell types, including those in the circumventricular organs, in the preoptic area of the hypothalamus, and muscle cells [66,67]. Overproduction of the aforementioned cytokines, especially IL-6 and APR are the cornerstones for the development of the symptoms observed in VL.
Table 1. Pathogenic mechanisms proposed to account for the clinical manifestations of VL deduced from organ/tissues and potential mediators involved in human patients and animal models.
| Clinical and Laboratory Manifestation | Model | Proposed Mediator | Organ and System Involved | Proposed Mechanism |
|---|---|---|---|---|
| Constitutional symptom | ||||
| Fever | Rodents (rabbit, mouse, rat, guinea pig) | IL-1β, IL-6, TNF-α | Circumventricular organs, hypothalamus, preoptic area, the central thermoregulatory circuitries | Exogenous and endogenous pyrogens cross the brain barrier and reach the hypothalamus, where prostaglandin A2 acts via the prostaglandin E receptor 3, generating fever |
| Weight loss | Rodents (mouse, rat) | IL-1β, IL-6, TNF-α, other cytokines | Circumventricular organs, hypothalamus, muscles, adipose tissue | “Sickness behavior” with loss of appetite, plus fever increasing the consumption of energy, sarcopenia due to IL-6, and acute phase response |
| Nausea and vomiting | Dog, cat, ferret, nonhuman primates, human | Unknown | Circumventricular organs, central-pattern generator, nucleus tractus solitarius. | Stimuli from the viscera transported by the blood arrive in the area postrema and by the abdominal vagal afferents to the nucleus tractus solitarius. Neurons project to a central-pattern generator, which coordinates the act of emesis, and project to the ventral medulla and hypothalamus, from which cortical brain areas are reached |
| Anemia | Mouse, dog, human | IL-6, IL-1β, hepcidin, ferroportin. Enlarged spleen | Bone marrow, liver, enterocytes, splenic macrophages. | IL-6 and Il-1b trigger the acute phase response protein hepcidin that inhibits the iron-exporter ferroportin, depriving bone marrow from iron. Also, phagocytosis by macrophages of the enlarged spleen |
| Localized symptom | ||||
| Hepatosplenomegaly | Dog, mouse, hamster, human | Hyperplasia, hypertrophy. | Spleen, liver | Proliferation of macrophages and amastigotes in spleen and liver |
| Edema | Dog, hamster, nonhuman primates, human | IL-6, IL-1β | Liver, blood vessels | Acute phase response diminishes the synthesis of albumin. Hypoalbuminemia decreases the oncotic pressure and plasma leaks to the interstitial space |
| Cough and dyspnea | Hamster, dog, cat, human | IL-6, IL-13, IL-4 | Pneumonitis in the alveolar interstitial space. Alveolar space | Interstitial inflammation and thickening alveolar space or bacterial pneumonia due to regulatory cytokines |
| Kidney failure | Hamster, mouse, dog, cat, human | Unknown, likely multicausal. Amyloid. Medication | Glomeruli, tubules | Polyclonal hypergammaglobulinemia secondary to IL-6 secretion, proteinuria leading to proximal tubular injury, associated with glomerular inflammation. Amyloid deposition. Drug toxicity |
| Liver involvement | Hamster, mouse, dog, human | Undetermined cytokines | Hepatocytes, bile ducts. Hepatic venules | Hepatocyte degeneration and death. Kupffer cells parasitized with amastigotes. Disseminated intravascular coagulation causing microthrombi. Fibrosis associated with Ito’s cells transformation into fibroblasts |
| Diarrhea | Hamster, mouse, dog, human | Parasitism. Malnutrition | Intestinal mucosa | Heavy parasitism with mucosal inflammation |
| Post-kala-azar dermal leishmaniasis | Human | IFN-γ, TNF-α, IL-12, IL-10 | Skin | Skin immunity is reactivated after cure, leading to a decrease in regulatory T cells, TGF-β, and IL-10 levels and an increase in IFN-γ, TNF-α, and IL-12 |
| Laboratory changes | ||||
| Neutropenia | Mouse, dog, nonhuman primates, human | Undetermined mediators | Bone marrow, spleen, liver vasculature | Parasitized and inflamed marginated pools of spleen, liver, and bone marrow may increase the disappearance of neutrophils by mechanisms such as necroptosis, pyroptosis, and neutrophil extracellular traps |
| Thrombocytopenia | Dog, hamster, nonhuman primates, human | IL-6, IL-1β, tissue-factor | Vasculature | IL-6, IL-1β, and TNF-α increase the expression of tissue factor, triggering the extrinsic pathway of coagulation and depleting coagulation factors, including platelets |
| Hyperglobulinemia | Dog, hamster, nonhuman primates, human | IL-6 | Liver, bone marrow | B-cells’ differentiation into antibody, producing plasma cells under the stimuli of IL-6 |
| Hypoalbuminemia | Dog, hamster, nonhuman primates, human | IL-6 | Liver | IL-6 induces acute phase reaction that reduces the synthesis of albumin |
| C-reactive protein | Dog, human | IL-6 | Liver | IL-6 induces acute phase reaction that increases the synthesis of C-reactive protein |
| Erythrocyte sedimentation rate | Human | IL-6, fibrinogen | Liver | IL-6 induces acute phase reaction that change the balance between pro- and antisedimentation factors |
| Haemophagocytic lymphohistiocytosis syndrome | Human | Cytokine storm, IL-6, IFN-γ, TNF-α, IL1β, IL-10 | Bone marrow, systemic | Activated CD8+ T cells and macrophages stimulate each other |
| Amyloidosis | Hamster, human | IL-6, serum amyloid A. | Kidneys, systemic. | IL-6 induces acute phase reaction that increases the synthesis of amyloid proteins that deposit in tissues |
| Complications and death | ||||
| Hemorrhage | Dog, hamster, human | IL-6, IL-1β, tissue-factor. | Vasculature | Leishmanial and bacterial sepsis: IL-6, IL-1β, and TNF-α increase the expression of tissue factor, triggering the extrinsic pathway of coagulation and consumption coagulopathy. Microthrombi can lead to multiorgan failure and death |
| Bacterial infections | Human | IL-10, IL-4, TGF-β. | Lungs, skin, urinary tract, blood | Regulatory cytokines, lymphopenia with lymphocyte apoptosis, following cytokine storm. Secondary malnutrition leads to several immunological defects. Bacterial sepsis further exacerbating leishmanial sepsis: disseminated intravascular coagulation causing microthrombi, which can lead to multiorgan failure, septic shock, and death |
The APR overexpressed by the hepatocytes include CRP (C-reactive protein), hepcidin, procalcitonin, CD14, serum amyloid A protein, fibrinogen, ferritin, and complement proteins C3 and C4 [68-70]. The different APRs produced vary in level. CRP is highly correlated with IL-6 [71] and can increase by a few thousand folds in VL, caused by both L. donovani and L. infantum [72-75]. Albumin decreases in level during the acute phase response, hence a negative APR, regularly notable in the sera of VL patients [58,76]. ESR (erythrocyte sedimentation rate) is almost always present at a high value in VL. This is a nonprotein APR whose level is known to change in response to plasma fibrinogen levels and plasma viscosity, hence an “indirect” APR [77-79]. APR rises with the increase in procoagulant and the decrease in anticoagulant proteins, fueling the already activated coagulation cascade, leading to fibrinolysis and bleeding [80].
Fever is one of the most notable symptoms of VL. It is mediated by the proinflammatory cytokines, called endogenous pyrogens, mostly IL-1β, TNF-α and IL-6. Endogenous pyrogens are synthesized and released after PRR (likely Toll-like receptor 2) interaction with Leishmania in macrophages [60,81]. After reaching the hypothalamus via the circumventricular organs that lack the blood–brain barrier, the pyrogens trigger the release of prostaglandin A2 via binding the prostaglandin E receptor in the preoptic area of the hypothalamus. This is the brain region where fever is generated in neurons within the central thermoregulatory circuitries [82,83].
Weight loss is also a common manifestation of VL, resulting from severe malnutrition [84,85]. Patients with VL-driven malnutrition are at higher risk of death [86-90]. While the mechanisms are unknown, there are at least three main possibilities to account for the weight loss, all linked to persistent systemic inflammation due to the production of proinflammatory cytokines, i.e.: (a) loss of appetite as part of the illness following systemic inflammation [67,91,92]; (b) fever that sharply increases the consumption of energy [93]; and (c) acute phase response that increases muscle catabolism leading to sarcopenia, thereby making amino acids abundantly available for APR synthesis in the liver [94,95,96]. The actions of TNF-α, IL-1β and especially IL-6 on the hypothalamus and muscles appear to play decisive roles in the development of cachexia.
Nausea and emesis are infrequently encountered symptoms of VL with multiple potential causes, including iatrogenic, toxic and infectious causes, gastrointestinal disorders and central nervous system or psychiatric dysfunctions [97]. Signals to cause these symptoms may be transmitted from the infected viscera with the blood flow to the area postrema, one of the circumventricular organs where the blood–brain barrier is absent and also by the abdominal vagal afferents to the nucleus tractus solitarius. Stimulated neurons then send signals to a central-pattern generator, which coordinates the act of emesis and to the ventral medulla and hypothalamus, reaching the cortical brain areas [98]. Vomiting has been reported to be associated with increased mortality of VL patients in both Africa and Brazil [58,86,88,89,90,99,100,101,102,103]. In these patients, it is associated with higher concentrations of IL-6 according to uni- and multi-variate analyses [104]. The chemical mediators of nausea and vomiting in VL are unknown. These are the alarm symptoms in dengue, which is, like VL, also associated with cytokine storm, involving IL6 and other proinflammatory cytokines [105]. Whether they are the mediators of nausea and vomiting awaits further investigation.
Anemia is also a common clinical manifestation and a risk factor for poor outcome of VL [58,86,88,89,90,100,101,102,103,106,107,108,109,110,111,112]. It is typically due to iron deficiency, with hypochromia and microcytosis marked by a low serum iron concentration [113]. However, the mechanical destruction of red cells, neutrophils and platelets alone in the enlarged spleen, e.g., hypersplenism contributes significantly to anemia, independent of inflammation [114]. Moreover, although most VL patients suffer from iron-deficient anemia [113], it is rarely caused by bleeding, indicative of the participation of a different mechanism in VL. This process may involve the APR of VL, hepcidin—an iron-regulating peptide. It functions to inhibit ferroportin, which mobilizes iron from all the iron-transporting cells (enterocytes, spleen and liver macrophages) into plasma for transport into bone marrow for hematopoiesis [115,116]. Indeed, anemia is negatively correlated with the level of IL-6, the cytokine that regulates APR, including hepcidin [104]; hepcidin mRNA is negatively correlated with hemoglobin in patients with VL [117]. In summary, while the destruction of red cells by hypersplenism is partially responsible for the anemia in VL, the APR contributes to this via the iron-depleting action of hepcidin under the control of proinflammatory cytokines, especially IL-6.
4. Localized Symptoms
Hepatosplenomegaly is one of the most important clues for diagnosing VL. It is determined by lymphoid hyperplasia, as histopathological studies have revealed that the spleen and the liver are full of parasites. There is no evidence of passive hypertensive congestion to explain splenomegaly. In spite of a marked atrophy of the splenic white pulp associated with necrosis and fibrosis of thymus-dependent areas, there is an accumulation of parasites-containing macrophages and plasma cell hyperplasia, accounting for the increase in the spleen volume. The number of parasites in the spleen and its architecture disruption is associated with the activities of proinflammatory and regulatory cytokines produced in diseased humans and dogs [118-121]. In both cases, parasite proliferation is promoted by the production of TGF-beta together with IL-10, produced locally by CD25(+)Foxp3(−) T cells and systematically by CD25(+)Foxp3(+) T cells [55,122,123]. The spleen increases in size to >10 times of its normal volume and appears to play a central role in the pathogenesis of VL. Splenectomy dramatically improves patients’ clinical conditions, although it does not cure the disease of HIV-co-infected patients [124]. Some degree of hypertrophy is also seen in the liver due to the proliferation of Kupffer cells in patients infected with either L. donovani or L. infantum [119,125].
Protracted edema (swelling) with or without anasarca (generalized edema) is a consequence of APR [109] and a risk factor for death in VL [58,87,88,89,90,101,102,103,107,108,109,111,126,127]. It is caused by a reduced concentration of albumin in plasma. Albumin is a negative APR and the principal protein determinant of oncotic plasma pressure. When it is low, plasma leaks to the interstitial space, leading to edema [128]. Therefore, hypoalbuminemia is VL’s primary cause of edema [68,69,70], consistent with the observation that IL-6 is elevated in edematous patients [104]. Hepatic failure is another cause of hypoalbuminemia, but this is rare in VL [76,119].
Cough is a common symptom and, together with the presence of pulmonary rales and dyspnea, indicates lung involvement in VL. This symptom is caused by a combination of alveolar bacterial pneumonia and interstitial proinflammatory response due to systemic Leishmania infection. These symptoms deserve prompt attention, since they are significantly associated with patients’ death, as found in many independent studies [58,89,101,102,103,109,110,111,126,129]. Interstitial pneumonitis is of inflammatory nature, as suggested by histopathologic studies that have demonstrated interstitial lung involvement where bacterial infection was absent; amastigotes were scarce, but Leishmania antigens were detected [130]. This conclusion is reinforced by an immunohistochemistry study of autopsy samples, showing interstitial accumulation of macrophages [131] and by computer tomography findings suggestive of association with respiratory symptoms [132]. The immunohistochemistry study also revealed a pattern of regulatory Th2 cytokines, indicative of an increased likelihood of bacterial superinfections. However, the histopathological and immunological data of VL suggest that the interstitial pulmonary involvement may be included in the category of profibrotic and proinflammatory interstitial lung diseases rather than bacterial infectious diseases [133,134,135].
Renal syndromes and low-grade renal failure were frequently observed in VL, and renal failure has been associated with increased mortality, as reported in some studies [89,109,127,136,137]. Proteinuria also reflects disease severity [58]. However, parasites are seldom seen in the kidneys. Renal involvement is related to interstitial nephritis and distinct glomerular participation, like collapsing segmental and focal glomerular sclerosis, necrotizing segmental and focal glomerular sclerosis and membranoproliferative lesion. The renal pathogenesis is not well understood and is likely multicausal. Due to the presence of polyclonal gamma globulins and IL-6 at high levels, it was hypothesized that proteinuria leads to proximal tubular injury, associated with glomerular inflammation [138,139]. AA amyloid glomerular deposits without mesangial hyperplasia have also been registered, likely as part of the APR [140]. However, the first suspected cause of interstitial nephritis is drug toxicity in VL, since most antileishmanial drugs are nephrotoxic [137].
Hepatitis is not uncommon in VL. The liver enzymes, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are slightly or moderately elevated, indicative of hepatocyte damage and useful as markers for disease severity [58,87,89,90,99,102,106,107,108,109,110,111,126,141,142]. Jaundice or an increased level of bilirubin is one of the most significant risk factors for death, particularly in patients with hemorrhages [12,58,87,89,90,99,101,102,103,107,108,109,110,111,127,129]. Many histopathology studies have demonstrated liver involvement, with ballooning degeneration of hepatocytes, Kupffer cells parasitized with amastigotes, chronic mononuclear infiltrate in the portal space, and fibrosis associated with Ito’s cells transformation into fibroblasts [119,125]. The association between liver inflammation with the elevation of IL-1β, TNF-α and IL-6 has been known in viral hepatitis, suggestive of a similar pathway to explain the liver involvement in VL [62,143]. However, liver involvement usually is not severe, as demonstrated by the rarity of hepatic encephalopathy. Researchers found that IL-6 was the serum cytokine with the strongest correlation, with an elevation of AST but not with that of ALT in a multivariate linear regression analysis [104]. As ALT is more specific to liver involvement than AST, the stronger association of AST with VL severity may reveal a more generalized, multiorgan involvement [144]. Indeed, multiorgan microthrombosis caused by disseminated intravascular coagulation (DIC), as seen in liver biopsies [125], may be another explanation for the rise of AST in VL.
Diarrhea is a prevalent symptom of VL caused by infection with L. infantum, and parasites are seen in the intestinal mucosa [126,145]. It is a consistent risk factor for death [58,86,87,88,89,90,99,100,101,102,103,106,107,108,109,111,127,146]. Severe malnutrition can lead to diarrhea through several mechanisms [147], potentially contributing to diarrhea seen in malnourished patients of VL. Since there is heavy parasitism with mucosal changes in patients with diarrhea and VL, it looks very likely that gut parasitism and local secondary inflammation are the leading cause of diarrhea in VL [126,148]. Comparison of gut mucosa of VL patients with and without diarrhea may further clarify the mechanism of its pathogenesis.
Post-kala-azar dermal leishmaniasis (PKDL) occurs in immunocompetent patients when infected by L. donovani, but only in patients with AIDS when infected by L. infantum [149,150]. This disparity cannot be explained fully without an in-depth study on the complexity of host–parasite–vector interactions, although the genetic differences between the two species are expected to play a role. At a time of a successful VL elimination program in South Asia, PKDL deserves renewed attention, since it is a source of parasites that fuels transmission and threatens elimination efforts. The lesions of South Asia PKDL caused by L. donovani are of the papulonodular and polymorphic type, with high parasite loads. Indian PKDL emerges in 5–10% of the VL patients 2–3 years after chemotherapy. They are rarely cured spontaneously. The East African type is monomorphic macular lesions with low parasite loads and patchy inflammatory infiltrates. East African PKDL emerges in 50–60% of the VL patients after or during the treatment, but 85% of the cases are self-healing. It has been proposed that PKDL occurs after cure of VL with reactivation of the specific immunity, i.e., a decrease in the levels of regulatory T cells, TGF-beta and IL-10, and an increase in those of IFN-γ, TNF-α and IL-12. L. donovani persisting in the skin may be detected by reactivated immune cells, which infiltrate into the cutaneous tissue, causing dermal inflammation by the secretion of IFN-γ, giving rise to dermal manifestations [19,151]. However, this conclusion clashes with the recent report that IL-10 remains elevated in PKDL [15]. Of relevance to mention is the report that treatment of VL with liposomal amphotericin B reduces the incidence of PKDL [152].
This entry is adapted from the peer-reviewed paper 10.3390/pathogens12070969
This entry is offline, you can click here to edit this entry!