Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Napabucasin (also known as BBI608) is a natural naphthoquinone originally identified as a cancer cell stemness inhibitor. Accumulated in vitro and in vivo evidence demonstrated that napabucasin showed significant anticancer effects in various types of cancers. Napabucasin showed multiple anticancer activities, including proliferation inhibition, cell death induction, cell cycle arrest, metastasis suppression, drug resistance overcoming, and stemness inhibition, etc., which were documented in many benchworks.
- napabucasin
- cancer
- STAT3
- NQO1
1. Cell Proliferation Inhibition and Cell Death Induction
Napabucasin inhibits cancer cell proliferation and growth of myeloid leukemia, liver, lung, ovarian, breast, B-cell lymphoma, osteosarcoma, glioblastoma, colorectal, prostate, biliary tract, pancreatic ductal, and hypopharyngeal cancer cells. Differential cytotoxicity of napabucasin was detected and observed after 6 h of treating different cell lines with increasing BBI608 concentrations [8]. In those cancer cell lines, the IC50s of napabucasin were typically at the μM or even nM level, which is substantially lower than the IC50s of several widely studied natural substances. In hepatocellular carcinoma, for example, napabucasin was far more active than cryptotanshinone in terms of anticancer activity [9].
Inducing cancer cell apoptosis is one of the general anticancer bases for napabucasin, and accumulated studies have demonstrated that napabucasin could activate intrinsic and extrinsic apoptosis pathways to cause apoptotic cell death. The cell viability of NHL (non-Hodgkin’s lymphoma) cell lines decreased with increasing concentrations of napabucasin, ranging from 0.001 to 2.0 μM for 48 h and 72 h, and, in DLBCL (diffuse large B-cell lymphoma) cells treated with napabucasin, the expression of cleaved caspase-3 and cleaved PARP significantly elevated in a concentration-dependent manner [10,11]. In addition, in drug-resistant lung cancer cells, napabucasin significantly induced cell apoptotic death when used as a single therapeutic agent [12]. In cisplatin-resistant small cell lung cancer (SCLC), napabucasin induces apoptosis by cleavage PARP and suppression of antiapoptotic proteins Mcl-1 and survivin [13].
2. Disruption of Cell Cycle
It is frequently maintained that the majority of cancer cells move through the cell cycle in an uncontrollable manner. Because of mutations that permit cell cycle advancement and impede exit, cancer cells keep dividing [23]. As a result of cancer cells’ higher reliance on cell cycle control regulatory pathways, there are opportunities to focus on disturbing the cell cycle, which represents a novel strategy for anticancer drug development. Abemaciclib, palbociclib, and ribociclib were approved by the FDA for breast cancer treatment as functioning as the CDK4/6 inhibitors that arrest the cell cycle in G1-phase [24]. Napabucasin owns such an ability as well, and it has been well documented in many experiments that napabucasin is a potent cell cycle arrest inducer. Of note, different from those singly disrupting one phase in a whole cell cycle, napabucasin could trigger cell cycle arrest in different cycle phases in cancer cells, dependent on the cancer type. For example, napabucasin attenuated the proliferation of glioma cancer cells by inducing G1/S-phase arrest in U87MG and LN229, along with its disruption of the cell cycle in CT26 colorectal cancer cells, and PC-3 and 22RV1 prostate cancer cells [16,18,25]. While in HGC-27 human gastric cancer cells, MDA-MB-231 breast cancer cells, and HeLa cervical cancer cells, napabucasin induces S-phase arrest [26] and induces G2/M phase arrest in HepG2.2.15 hepatic carcinoma cells [9]. Consistent with its ability to perform in breast cancer cells, after 24 h of dosing, napabucasin caused the cell cycle of H146 and H446 cells to arrest in the S-phase [13]. Accumulation in the percentage of cycling Molm13 acute myeloid leukemia cells in the G0/G1 phase and a significant reduction in the number of cells cycling in the S-phase were observed clearly after administration of napabucasin at increasing dosages [19]. Such alteration of the proportion between those two phases was detected, as well, in the U87MG cell line and LN229 cell line with increasingly introducing napabucasin [16]. This G1 phase arrest induced by napabucasin might be mediated by the upregulation of P21 and a reduction in Cyclin D1 and CDK4 in Molm13 cells and U87MG and LN229 cells [16,19].
3. Suppress Metastasis and Improve Drug Resistance
One of the main causes of cancer patients’ mortality is tumor metastasis, which accounts for about 90% of cancer-related deaths. The process of metastasis is the migration of cancer cells from their originating site to gradually colonized distant organs [20]. From the results of wound healing assays, transwell migration, and invasion assays on EOC20 cell lines after 24 h of the administration of napabucasin, fewer migrating SKOV3 ovarian cancer cells and A2780 ovarian cancer cells began to migrate as the drug concentration increased [27]. Additionally, better than ReoT3D and CPT-11 in monotherapy, napabucasin led to more antimigration action on CT26 mouse colon cancer cells [25]. Similar inhibited migration and invasion were also observed in U87MGc cells and LN229 cells and possibly by suppressing matrix metalloproteinase-2 (MMP2) and MMP9 [16], which could decompose the extracellular matrix (ECM) and stimulate cell invasion and metastasis through epithelial–mesenchymal transition (EMT) in vitro and in vivo. However, the detailed mechanisms remain unclear.
The effect of napabucasin on drug resistance is mentioned in a few studies. In nonsmall cell lung cancer (NSCLC) cells, napabucasin not only inhibits cell proliferation of cisplatin-resistant NSCLC cells alone but also enhances the cytotoxicity of cisplatin in drug-resistant sublines. As an independent treatment, napabucasin dramatically reduced the ability of all cisplatin-resistant sublines to proliferate in comparison to untreated cells. Napabucasin dramatically reduced cell proliferation in all resistant NSCLC sublines that represented each histological subtype of this cancer type when combined with cisplatin [12]. This finding suggests that napabucasin should be researched further as a novel drug for resensitizing NSCLC cells to cisplatin’s cytotoxic effects. Besides those resistant to cisplatin, napabucasin stimulates ovarian cancer cell death by enhancing its sensitivity to paclitaxel both in vitro and in vivo [27].
4. Inhibition on Cancer Stemness
Cancer stem cells (CSCs), also known as tumor-initiating cells, are a small subset of cells within a heterogeneous tumor that have the unique ability to initiate new cancer upon transplantation and share characteristics with embryonic and stromal stem cells, such as the ability to self-renew (divide to make more stem cells without transforming into a specialized cell) and differentiate (transform into a specialized cell) [28]. Such CSCs are usually considered the key factor in cancer development, relapse, drug resistance, and metastasis [21,29]. Napabucasin was originally demonstrated to inhibit cancer stem cell proliferation and stemness properties as evidenced by the inhibition of sphere formation and clonogenic growth in malignant gliomas cancer cells, hepatic cancer cells, prostate cancer cells, and biliary tract cancer cells [9,14,16,18,30]. In PCa stem cell stemness, napabucasin treatment dropped the protein expression level of Nanog, Klf4, survivin, C-myc, and β-catenin in PrCSCs in a dose-dependent manner. The considerably decreased mRNA expression of Nanog, Klf4, survivin, and β-catenin in napabucasin treatment was observed through qRT-PCR [18]. Such decreased levels of the stemness marker indicate the inhibitive effect of napabucasin on cell stemness. More importantly, although well performed in cancer cells, napabucasin did not appear to have adverse effects on hematopoietic or other normal adult stem cells. Besides these, the drug-resistant cancer cells showed stronger stemness properties, such as higher sporogenesis ability, colony formation ability, aldehyde dehydrogenase activity, and expression of stemness key factor [31]. One example is in MCF7/MCF7-R breast cancer cells with tamoxifen resistance. Behaving as the canonical STAT3 inhibitor, napabucasin attenuated the stemness of breast cancer stemness [32] and another type of cancer [30] and decreased stemness marker expression, colony formation ability, and aldehyde dehydrogenase (ALDH) activity of MCF7-R, which exhibited a higher stemness to MCF7 cells. As such, napabucasin reverses the tamoxifen resistance of MCF7/MCF7-R breast cancer cells and enhances its sensitivity toward tamoxifen [32].
Several signal pathways are involved in the regulation of cancer stem cell proliferation and differentiation, such as the STAT3, Wnt, and PI3K/Akt pathways [22]. Napabucasin inhibits cancer stemness mainly by inhibiting STAT3 and its target genes. Napabucasin kills cancer stem cells and decreases stem-cell-related biomarkers Nanog, SOX2, Oct4, CD90, Klf4, survivin, c-Myc, and β-catenin in liver cancer [9], breast cancer [32], prostate cancer [18], and so on. Napabucasin blocked the survival and self-renewal of cancer stem cells and inhibited their related proteins and gene expression in pharynx squamous cell carcinoma FaDu cells. In contrast, some regular chemical drugs or kinase-targeted drugs induced cancer stem-cell-related gene expression [30].
5. In Vivo Evidence
The anticancer effect of napabucasin has been confirmed in various animal models. As a monotherapy, napabucasin administration reduced tumor volume and inhibited tumor growth in an orthotopic tibial osteosarcoma model, an orthotopic glioma model, an HCC homograft mice model, and human acute myeloid leukemia xenograft murine models [9,11,16,19]. It also significantly prolonged the life span of tumor-burden mice in glioma and melanoma mice models [16,33]. Furthermore, napabucasin treatment alleviated bone osteolysis and bone destruction in a 4-week-old female BABL/c nude mice model [11]. In addition, napabucasin administration inhibited metastasis in an osteosarcoma nude mice model, a colon cancer cells liver metastasis model, and a pancreatic cancer xenograft model [11,30].
The in vivo anticancer effect of napabucasin mainly depends on the inhibition of stem cells, cell proliferation, and induction of apoptosis. Napabucasin downregulates stemness-related genes Nanog, SOX2, Klf4, and Oct4 CD90 and EpCAM mRNA expression levels both in tumor cells and tumor tissues [9]. Napabucasin treatment decreases Ki67-positive cells and increases apoptotic cells, suggesting that napabucasin inhibits cancer cell proliferation and triggers apoptosis [9,10].
Combination therapy of napabucasin with other drugs shows a synergistic anticancer effect. Napabucasin synergized with doxorubicin, docetaxel, and paclitaxel in a DLBCL diffuse large B-cell lymphoma xenograft model [10], prostate cancer xenograft models [18], and a model of peritoneal tumor of ovarian cancer [27], respectively. The undergoing mechanism of its synergistic potential could be its inhibition of cancer cell stemness or enhancement of certain anticancer medications, simultaneously performing its apoptosis-inducing ability.
6. Molecular Targets
STAT3, an oncogenic transcription factor, is a well-known promising anticancer target [36]. It has been verified as being essential in controlling the antitumor immune response because it serves as a point of convergence for many oncogenic signaling pathways. It is widely hyperactivated in both cancer and noncancerous cells within the tumor ecosystem and plays significant roles in inhibiting the expression of essential immune activation regulators and promoting the production of immunosuppressive factors. Among STAT protein family members, STAT3 is involved in numerous biological processes, including cell proliferation, survival, differentiation, and angiogenesis. Several inhibitors and their analogs of STAT3 have been well testified as effective anticancer drugs, such as Rapamycin, Angoline, Shikonin, and so on [37,38,39]. As mentioned above, the inhibitory effect of napabucasin on stemness is mediated by STAT3 [16]. With this, napabucasin is enabled to perform those properties listed above.
However, recent evidence showed that NQO1, a two-electron reductase involved in the detoxification of quinones and the bioactivation of certain quinones [40], was a major determinant of napabucasin’s efficacy. In those solid tumors with high expression of NQO1, such as lung cancer, and ovarian cancer, NQO1 was regarded as the therapeutic target due to its promotion of cancer cell growth at the early stage of carcinogenesis by binding and stabilizing the mutant or wild-type p53 and then inhibiting its degradation [41]. Napabucasin is an NQO1 substrate and selectively kills NQO1 high-expression cancer cells. In particular, the inhibitory effect of napabucasin on STAT3 was found to be mediated by NQO1-derived reactive oxygen species (ROS) [8]. It is reasonable that NQO1 acts as a target for napabucasin from a chemical point of view. Napabucasin has a naphthoquinone structure, and many compounds with this structure, such as β-lapachone and 2-methoxy-6-acetyl-7-methyljuglone (MAM), have been found to exert antitumor activity by targeting NQO1 [41,42]. Thus, napabucasin might act as an NQO1 bioactivatable drug. Consistent with this is the excessive ROS generation induced by napabucasin in NQO1 high-expression cells [26]. However, for those cells with low expression of NQO1, napabucasin treatment still caused the accumulation of ROS, which suggests that other targets and mechanisms should be involved in this process. Indeed, although napabucasin binds cytochrome P450 oxidoreductase with a weaker affinity than that of NQO1, once binding, it could also induce ROS generation [8], and the subsequent events occurred. As NQO1 activation triggers nonapoptotic cell death termed noptosis [43], the cell death induced by napabucasin may be cell-type-dependent. With this idea, as mentioned above, the anticancer activity on disrupting the cell cycle differing in cancer cells from type to type probably depends on different CDK targets or others.
Besides STAT3 and NQO1, napabucasin is identified to be an inhibitor of thioredoxin reductase 1 (TrxR1). It acts as a redox cycling substrate of TrxR1 and irreversibly inhibits cellular TrxR1 activity as a few STAT3 inhibitors [44]. Recently, aldehyde dehydrogenase was proposed to be another napabucasin target using thermal proteome profiling in whole zebrafish embryo lysate [45]. The anticancer molecular mechanisms of napabucasin are summarized in Figure 1.
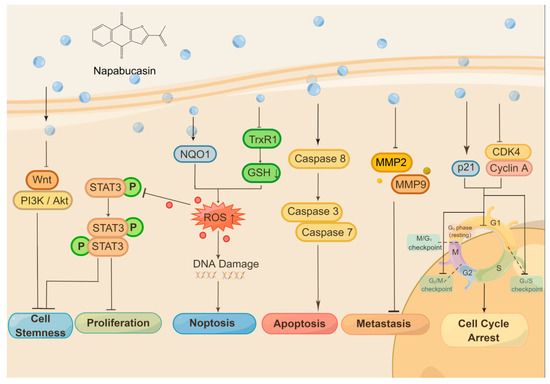
Figure 1. The anticancer molecular mechanisms of napabucasin.
Napabucasin shows great anticancer activity via different molecular mechanisms. Napabucasin itself inhibits several signaling pathways, such as Wnt, PI3K/Akt, and STAT3, and stands the inhibition of cancer cell stemness as one consequence, another as inhibition of cancer cell proliferation. Inhibition of STAT3 through ROS generation is mediated by NQO1, as napabucasin is an NQO1 substrate. Napabucasin is also identified as a TrxR1 inhibitor. The activation of the caspase 3/7/8 induced by napabucasin drives the cancer cell to apoptosis. In contrast, ROS generation triggers both STAT3 inhibition and DNA damage and finally induces noptosis, a nonapoptotic cell death. Meanwhile, napabucasin inhibits MMP2/MMP9, which takes the suppression of cancer metastasis as a result. In addition, napabucasin can disrupt the cell cycle on different phases or checkpoints in cancer cells from type to type. Figure 1 was drawn by Figdraw (ID:UIISU46533).
This entry is adapted from the peer-reviewed paper 10.3390/molecules28155678
This entry is offline, you can click here to edit this entry!