Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Obstetrics & Gynaecology
|
Oncology
Ovarian cancer (OC) is characterized by silent progression and late-stage diagnosis. It is critical to detect and accurately diagnose the disease early to improve survival rates. Tumor markers have emerged as valuable tools in the diagnosis and management of OC, offering non-invasive and cost-effective options for screening, monitoring, and prognosis.
- ovarian cancer
- cancer biomarkers
- early detection
- prognosis
- cancer diagnosis
1. Introduction
1.1. Background of Ovarian Cancer
In the United States in 2020, there were 1,806,590 new cases of cancer and 606,520 cancer-related deaths. Specifically, OC appears as the leading cause of death for female reproductive tract cancers. It is estimated that in 2020, there were 21,750 new cases and 13,940 deaths related to OC. Postmenopausal women are considered at high risk of developing OC because the likelihood of developing an advanced stage disease increases with age. An absence of early detection methods and the limited effectiveness of standard chemotherapy are the main factors contributing to this vulnerability [1]. The prevalence of OC among post-menopausal women is estimated to be 1 in 2500 [2]. OCs are diagnosed at an advanced stage for around 70% of the cases, resulting in a 5-year survival rate of only 30%. Nevertheless, the 5-year survival rate can exceed 90% when OC is detected early and confined to the ovaries. It is essential to gain a deeper understanding of the molecular causes of OC, even though, in the last 25 years, modest improvements in survival rates have been observed. Crucially, new biomarkers could aid in the early detection pathway, especially as less than 20% of OCs are diagnosed at a localized stage [3].
Among gynecological malignancies, malignant epithelial tumors (carcinomas) are the deadliest forms of OC. Currently, the classification of ovarian epithelial tumors has solely relied on the morphology of tumor cells. Six to nine cases per 100,000 women is the global incidence rate of these cancers [4]. Broadly, there are three categories of OC based on the types of ovarian cells involved. Surface epithelial cells are the cell type in category one, and can cover the ovary and be subdivided into many subtypes. The second category consists of germ cells, which are the cells that ultimately develop into ova. OC subtypes related to germ cells encompass yolk sac tumors, immature teratoma, and dysgerminoma. Finally, sex cord–stromal cells comprise the third category. These tumors include malignant granulosa cells and Sertoli–Leydig cells [5,6].
Considering their histopathology and molecular genetic alterations, the subtypes of epithelial ovarian cancers (EOCs) are as follows: (1) high-grade serous, (2) endometrioid, (3) clear cell, (4) mucinous, and (5) low-grade serous carcinomas. EOCs constitute more than 95% of all OC cases (Figure 1). Moreover, based on characteristics such as extent of cell proliferation, the presence of nuclear atypia, and stromal invasion, the tumors are further subdivided as benign, borderline (intermediate), and malignant (carcinoma). This detailed categorization aids in providing a comprehensive understanding of the nature and behavior of these tumors [7,8,9].
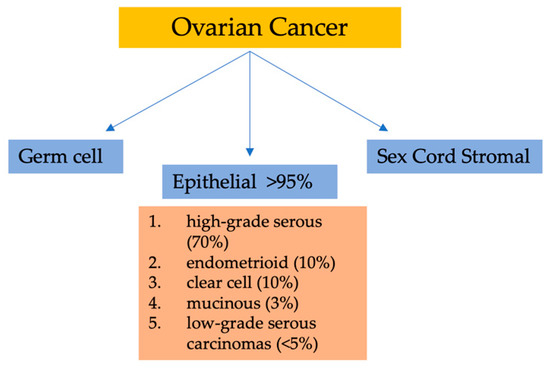
Figure 1. Classification of ovarian malignancies.
The dualistic model of ovarian carcinogenesis encompasses the primary histopathological subtypes, consolidating them into two distinct categories—type I and type II—based on clinical, genetic, and developmental characteristics. In terms of diagnosis, type I ovarian carcinomas constitute approximately 30% of cases, whereas type II tumors represent the majority, accounting for about 70%. Type I tumors tend to be confined to the ovaries (stage I) and generally exhibit a more favorable prognosis, contributing to a mere 10% of ovarian cancer-related fatalities. Type I tumors correspond to an initial phase characterized by clinically less aggressive behavior. These tumors typically include low-grade clear-cell, serous, mucinous, and endometrioid subtypes, with rare occurrences of seromucinous and Brenner tumors. Conversely, type II tumors display a more aggressive nature and account for the majority of cases of epithelial ovarian cancers (EOCs); they are typically diagnosed at advanced stages (III and IV), leaving limited prospects for a cure. This category comprises high-grade undifferentiated, endometrioid, serous, and malignant mixed mesodermal tumors, which are associated with poorer prognosis and clinical outcomes. Astonishingly, type II tumors are responsible for an overwhelming 90% of ovarian cancer-related deaths. Consequently, some experts advocate directing extensive screening efforts towards type II tumors as a strategic approach to potentially yield substantial improvements in patient outcomes [10,11,12,13,14].
1.2. Significance of Early Diagnosis in Ovarian Cancer
OC is a complicated and diverse group of diseases characterized by variations in morphology and biological behavior. Although its prevalence is less than that of breast cancer, the impact of OC is disproportionally higher with a significant number of deaths attributed to the disease. OC proves fatal for the vast majority of patients diagnosed with advanced (stage III) ovarian tumors since recurrence following surgery and chemotherapy is seen in around 75% of cases. Globally, OC is considered as the most lethal gynecological cancer and the fifth most common cause of cancer-related deaths among women in the Western world [8,9,15]. By improving the efficacy of screening methods, such as tests for specific biomarkers, the chances of detecting OC at an early stage could be increased.
2. MicroRNAs
MicroRNAs (miRNAs) are short RNA molecules that regulate gene expression and play crucial roles in various biological processes. The production and way of action of miRNA is displayed in Figure 2. MiRNA genes are transcribed to produce pri-miRNA, which is cleaved to form pre-miRNA. In the cytoplasm, pre-miRNA is further cleaved to generate a miRNA duplex. The mature miRNA regulates gene expression by targeting mRNA for cleavage or translation repression based on miRNA–mRNA complementarity. Their dysregulation is associated with numerous human conditions, including cancer [88]. Aberrant miRNA expression in ovarian cancer has diagnostic and prognostic potential as circulating miRNAs (cirMiRs), offering non-invasive biomarkers [89]. MiRNAs regulate gene expression by targeting multiple genes, making them valuable for understanding gene behavior [90,91]. MiRNAs are stable in the circulation, bound tochaperone protein Argonaute 2(Ago2) or enclosed in extracellular vesicles, resisting degradation by ribonucleases [92,93]. Over 2500 miRNAs have been identified, capable of targeting multiple genes within pathways, providing valuable insights into gene behavior [90,91,94]. Let-7 and miR-200 miRNA families are implicated in OC development. Let-7 has potential for selecting chemotherapy, while miR-200’s role in chemo-sensitivity is uncertain. miRNAs hold promise as chemotherapy response predictors, but further validation is needed. Plasma/serum miRNAs offer potential for early OC diagnosis, but before clinical utility additional research is required [94]. Dysregulated miRNAs in OC act as tumor suppressors or oncogenes. Low expression of miR-processing enzymes is linked to advanced tumor stage and poor outcomes. Let-7 and miR-200 families are frequently altered in OC. Various miRNAs have diagnostic and therapeutic potential. Serum miRNA panels show promise for OC diagnosis and monitoring. miRNAs are valuable tools for OC management [10]. Aberrant miRNA expression in OC is associated with chemoresistance, including let-7e, miR-30c, miR-125b, miR-130a, miR-335, miR-340, miR-381, and miR-520f [95].
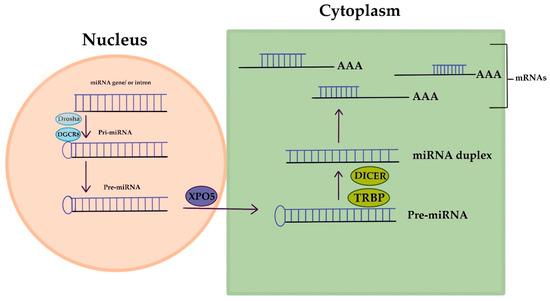
Figure 2. MicroRNA’s creation and way of action.
MiRNA expression profiles show diagnostic potential in ovarian cancer, with specific miRNAs differentially expressed in OC samples. Circulating miRNAs in blood and urine are promising diagnostic markers. MiRNAs also correlate with histotypes, chemoresistance, and prognosis, offering insights into disease progression and chemotherapy outcomes in OC [96]. Altered miRNA expression in OC correlates with disease stage, treatment response, and overall survival. miR-21, miR-200a, and miR-200c have diagnostic and prognostic value, while let-7f and miR-141 are associated with worse progression-free survival. miR-193a acts as a tumor suppressor [89]. In an enlightening study by Yokoi et al., the discrimination of early-stage ovarian cancers from benign tumors was achieved with an impressive sensitivity of 86% and specificity of 83% by employing a panel of eight miRNAs. Furthermore, the presence of miRNAs was detected in EVs isolated from cultured ovarian cancer cell lines [97].
3. DNA Methylation Patterns
DNA methylation markers offer early detection potential in OC, unlike CA125 [98,99]. The utilization of cell-free DNA (cfDNA) methylation markers shows promise in identifying early-stage OC patients within the average-risk population. Since early-stage OC patients typically remain asymptomatic, incidental diagnosis is common. Consequently, the development of effective DNA methylation markers holds promise for early OC detection and requires continued investigation to enhance clinical applicability [100,101]. Frequent genetic alterations in cancer involve hypermethylation of tumor suppressor promoters and hypomethylation of oncogenes. Methylation-specific PCR (MSP) stands as a highly sensitive technique, capable of detecting one methylated allele among 1000 unmethylated alleles. The occurrence of promoter hypermethylation escalates as the disease progresses [102,103].
Multiplexed methylation-specific PCR (MSP) of cfDNA for seven genes showed high sensitivity (85%) and specificity (91%) for early-stage ovarian cancer compared to CA125 alone [104]. Widshwendter et al. developed a three-DNA-methylation-serum-marker panel using targeted ultra-high coverage bisul-fite sequencing. The panel successfully differentiated high-grade serous ovarian cancer patients from healthy women or those with benign pelvic masses, achieving a sensitivity of 41.4% and specificity of 90.7%. When applied to serum samples collected 1–2 years before ovarian cancer diagnosis, the methylation panel showed a sensitivity of 16.7% and specificity of 96.9% [98].
DNA methylation is a useful marker for cancer cell fraction analysis, providing advantages in terms of time, cost, and independence from allelic status. This approach, along with other markers, reduces reliance on pathologists and enables efficient analysis of ovarian cancer cell fractions [105]. A study identified DNA methylation markers (COL23A1, C2CD4D, and WNT6) with high sensitivity and specificity for early ovarian cancer (OC) detection. The markers exhibited aberrant methylation patterns in early-stage OC and showed promise in discriminating OC from healthy individuals. The panel demonstrated potential as a complementary approach for early OC diagnosis, particularly in CA125-negative samples [98]. Late-stage methylation markers show limited utility in early-stage ovarian cancer (OC) detection, while early-stage markers demonstrate satisfactory discrimination. Hypomethylated regions display reversal to baseline levels in late-stage OC. Early-stage methylation markers remain stable during cancer progression, offering potential for OC detection across all stages [106]. SIM1 and ZNF154 genes were identified as potential methylation markers for ovarian cancer cell fraction estimation. ZNF154 was validated as a reliable marker, offering a cost-effective and efficient method for assessing ovarian cancer cell fraction using pyrosequencing [107].
Ovarian clear cell carcinoma (OCCC) can be classified into two distinct clusters based on DNA methylation patterns. Cluster 1 is associated with advanced stage, poorer outcomes, TP53 mutation, and macroscopic residual disease, while Cluster 2 is characterized by early stage, aneuploidy, ARID1A/PIK3CA mutation, and longer overall survival. Immune-related pathways and ARID1A mutations contribute to the molecular and clinical heterogeneity of OCCC [108]. Ovarian cancer DNA methylation analysis identified 250 prognosis-related loci, revealing six subtypes with distinct patterns and prognoses. Subtype 2 had the highest methylation and best prognosis, while subtypes 4 and 5 had lower methylation and poor prognoses. Hypomethylation correlated with worse outcomes. These subtypes could serve as biomarkers for personalized treatment and prognosis prediction [109]. A study identified 89 CpG sites associated with epithelial ovarian cancer (EOC) risk, including 12 CpG sites and five genes (MAPT, HOXB3, ABHD8, ARHGAP27, and SKAP1) showing consistent associations. Methylation at these sites may regulate gene expression and influence EOC risk, particularly for serous and high-grade serous ovarian cancer. Integration of genetic, methylation, and gene expression data provides insights into EOC development and potential personalized treatment targets [110]. HOXA10 and HOXA11 genes show significant DNA methylation differences in ovarian cancer, with HOXA11 methylation associated with poor prognosis and residual tumor. HOXA10 methylation is higher in poorly differentiated cancers. Low HOXA11 methylation correlates with minimal residual tumor and serves as an independent prognostic marker. Methylation frequency increases from non-neoplastic to primary ovarian cancer, highlighting their diagnostic and prognostic potential [111].
Comprehensive analysis revealed (hypomethylated-upregulated) HOUP genes associated with ovarian cancer progression and potential prognostic markers, while (hypermethylated-downregulated) HEDW genes were enriched in cancer-related pathways. Dysregulated hub genes and negative correlations with methylation levels were identified, providing insights into ovarian cancer epigenetic alterations and biomarkers [112]. Cervical scrapings from Pap tests showed significant hypermethylation in five genes in OC patients. An integrated model incorporating methylation levels predicted OC risk with high sensitivity and specificity, offering potential for enhanced detection of female genital tract malignancies [113]. cfDNA methylation analysis identified specific differentially methylated regions (DMRs) associated with OC. A customized methylation panel revealed OC-specific DMRs with distinct methylation patterns, suggesting their potential as diagnostic and prognostic markers [114]. Finally, in a comprehensive analysis a validated serum marker panel using targeted bisulfite sequencing showed high sensitivity and specificity, suggesting the potential of DNA methylation patterns for early OC detection [98].
4. Circulating Tumor Cells
Circulating tumor DNA (ctDNA) allows non-invasive detection of ovarian cancer mutations, such as PIK3CA and KRAS, with potential as diagnostic and prognostic markers in liquid biopsy. Differentiated from lymphocyte DNA, cfDNA exhibits characteristic fragmented size [115]. Research efforts have primarily focused on analyzing the fraction of cfDNA originating from tumors, known as circulating tumor DNA (ctDNA) [116]. ctDNA is primarily released from tumor cells through apoptosis [117,118]. Recent advancements in deep sequencing and droplet digital PCR (ddPCR) techniques have enabled the detection of specific mutations, loss of heterozygosity (LOH), DNA hypermethylation, copy number variations, and even the presence of single nucleotide variants in minute quantities of ctDNA [119,120,121,122,123].
Swisher et al. detected tumor-specific TP53 mutations in cfDNA using traditional PCR, with a 30% detection rate in plasma or serum samples. ctDNA analysis shows potential as a non-invasive method for identifying cancer-specific mutations across different stages [124]. Advanced sequencing technologies, such as tagged amplicon sequencing (TAm-Seq) and duplex sequencing, enhance ctDNA detection with high sensitivity (as low as 2% allelic fractions) and specificity (97% for TAm-Seq). However, the applicability in early-stage cancers and the balance between sensitivity and specificity require further investigation. Duplex sequencing reveals low-level mutant TP53 events in peritoneal fluid, suggesting normal physiological processes involve mutant TP53 [125,126,127,128]. The integration of ctDNA with CA125 in a multi-cancer investigation achieved a high sensitivity of 98% for detecting ovarian cancer, primarily in advanced-stage tumors [129]. PIK3CA and KRAS mutations in ctDNA serve as prognostic markers and indicate outcomes in epithelial ovarian cancer patients. ctDNA detection rates correlate with advanced stage and peritoneal cytology, while ctDNA presence at primary treatment predicts shorter recurrence-free survival [130].
ctDNA detection in EOC patients correlates with advanced stages, high-grade disease, disease progression, and higher mortality rates. CtDNA outperforms CA-125 as a prognostic indicator for recurrence, with its presence after surgery strongly associated with reduced recurrence-free survival. CtDNA provides valuable insights for risk assessment and monitoring of EOC recurrence. Genomic profiling reveals frequent mutations in TP53, ARID1A, KRAS, and PIK3CA [131]. Finally, a systematic review of eight studies involving 627 ovarian epithelial cancer patients found that ctDNA is significantly associated with decreased overall survival (OS) and progression-free survival (PFS). Serum-derived ctDNA showed a strong relationship with reduced OS, while plasma-derived ctDNA had some heterogeneity. The analysis also indicated that ctDNA could serve as an independent risk factor and a potential biomarker for evaluating ovarian cancer prognosis [132].
This entry is adapted from the peer-reviewed paper 10.3390/life13081689
This entry is offline, you can click here to edit this entry!