(BEN)-Domain containing protein 3 (BEND3) is a transcription factor that plays a critical role in the regulation of gene expression in mammals. While there is limited research on the role of BEND3 as a tumor suppressor or an oncogene and its potential role in cancer therapy is still emerging, several studies suggest that it may be involved in both the processes. Its interaction and regulation with multiple other factors via p21 have already been reported to play a significant role in cancer development, which serves as an indication of its potential role in oncogenesis. Its interaction with chromatin modifiers such as NuRD and NoRC and its role in the recruitment of polycomb repressive complex 2 (PRC2) are some of the additional events indicative of its potential role in cancer development.
- BEND3
- tumor suppressor
- oncogenic driver
1. Introduction
2. BEND3 Structure and Its Interaction with DNA
3. BEND3-Mediated Chromatin Regulation
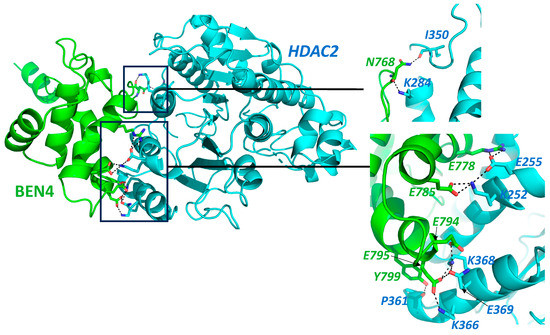
This entry is adapted from the peer-reviewed paper 10.3390/cancers15143685
References
- Ablett, M.P.; Singh, J.K.; Clarke, R.B. Stem Cells in Breast Tumours: Are They Ready for the Clinic? Eur. J. Cancer. 2012, 48, 2104–2116.
- Witt, A.E.; Lee, C.-W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a Cancer Stem Cell-Specific Function for the Histone Deacetylases, HDAC1 and HDAC7, in Breast and Ovarian Cancer. Oncogene 2017, 36, 1707–1720.
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241.
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830.
- Kurniawan, F.; Chetlangia, N.; Kamran, M.; Redon, C.E.; Pongor, L.; Sun, Q.; Lin, Y.-C.; Mohan, V.; Shaqildi, O.; Asoudegi, D.; et al. BEND3 Safeguards Pluripotency by Repressing Differentiation-Associated Genes. Proc. Natl. Acad. Sci. USA 2022, 119, e2107406119.
- Zhang, J.; Zhang, Y.; You, Q.; Huang, C.; Zhang, T.; Wang, M.; Zhang, T.; Yang, X.; Xiong, J.; Li, Y.; et al. Highly Enriched BEND3 Prevents the Premature Activation of Bivalent Genes during Differentiation. Science 2022, 375, 1053–1058.
- Khan, A.; Giri, S.; Wang, Y.; Chakraborty, A.; Ghosh, A.K.; Anantharaman, A.; Aggarwal, V.; Sathyan, K.M.; Ha, T.; Prasanth, K.V.; et al. BEND3 Represses RDNA Transcription by Stabilizing a NoRC Component via USP21 Deubiquitinase. Proc. Natl. Acad. Sci. USA 2015, 112, 8338–8343.
- Khan, A.; Prasanth, S.G. BEND3 Mediates Transcriptional Repression and Heterochromatin Organization. Transcription 2015, 6, 102–105.
- Sathyan, K.M.; Shen, Z.; Tripathi, V.; Prasanth, K.V.; Prasanth, S.G. A BEN-Domain-Containing Protein Associates with Heterochromatin and Represses Transcription. J. Cell Sci. 2011, 124, 3149–3163.
- Pitchai, G.P.; Kaulich, M.; Bizard, A.H.; Mesa, P.; Yao, Q.; Sarlos, K.; Streicher, W.W.; Nigg, E.A.; Montoya, G.; Hickson, I.D. A Novel TPR-BEN Domain Interaction Mediates PICH-BEND3 Association. Nucleic Acids Res. 2017, 45, 11413–11424.
- Lai, A.Y.; Wade, P.A. Cancer Biology and NuRD: A Multifaceted Chromatin Remodelling Complex. Nat. Rev. Cancer 2011, 11, 588–596.
- Barghout, S.H.; Aman, A.; Nouri, K.; Blatman, Z.; Arevalo, K.; Thomas, G.E.; MacLean, N.; Hurren, R.; Ketela, T.; Saini, M.; et al. A Genome-Wide CRISPR/Cas9 Screen in Acute Myeloid Leukemia Cells Identifies Regulators of TAK-243 Sensitivity. JCI Insight 2021, 6, e141518.
- Khan, A.; Prasanth, S. BENDing with Polycomb in Pluripotency and Cancer. BioEssays 2003, 2300046.
- Zheng, L.; Liu, J.; Niu, L.; Kamran, M.; Yang, A.W.H.; Jolma, A.; Dai, Q.; Hughes, T.R.; Patel, D.J.; Zhang, L.; et al. Distinct Structural Bases for Sequence-Specific DNA Binding by Mammalian BEN Domain Proteins. Genes Dev. 2022, 36, 225–240.
- Abhiman, S.; Iyer, L.M.; Aravind, L. BEN: A Novel Domain in Chromatin Factors and DNA Viral Proteins. Bioinformatics 2008, 24, 458–461.
- Shiheido, H.; Shimizu, J. Basic Amino Acid Residues Located in the N-Terminal Region of BEND3 Are Essential for Its Nuclear Localization. Biochem. Biophys. Res. Commun. 2015, 457, 589–594.
- Xie, W.; Ling, T.; Zhou, Y.; Feng, W.; Zhu, Q.; Stunnenberg, H.G.; Grummt, I.; Tao, W. The Chromatin Remodeling Complex NuRD Establishes the Poised State of RRNA Genes Characterized by Bivalent Histone Modifications and Altered Nucleosome Positions. Proc. Natl. Acad. Sci. USA 2012, 109, 8161–8166.
- Harikumar, A.; Meshorer, E. Chromatin Remodeling and Bivalent Histone Modifications in Embryonic Stem Cells. EMBO Rep. 2015, 16, 1609–1619.
- dos Santos, R.L.; Tosti, L.; Radzisheuskaya, A.; Caballero, I.M.; Kaji, K.; Hendrich, B.; Silva, J.C.R. MBD3/NuRD Facilitates Induction of Pluripotency in a Context-Dependent Manner. Cell Stem Cell 2014, 15, 102–110.
- Sims, J.K.; Wade, P.A. Mi-2/NuRD Complex Function Is Required for Normal S Phase Progression and Assembly of Pericentric Heterochromatin. Mol. Biol. Cell 2011, 22, 3094–3102.
- Kaji, K.; Caballero, I.M.; MacLeod, R.; Nichols, J.; Wilson, V.A.; Hendrich, B. The NuRD Component Mbd3 Is Required for Pluripotency of Embryonic Stem Cells. Nat. Cell Biol. 2006, 8, 285–292.
- van den Berg, D.L.C.; Snoek, T.; Mullin, N.P.; Yates, A.; Bezstarosti, K.; Demmers, J.; Chambers, I.; Poot, R.A. An Oct4-Centered Protein Interaction Network in Embryonic Stem Cells. Cell Stem Cell 2010, 6, 369–381.
- Zhu, D.; Fang, J.; Li, Y.; Zhang, J. Mbd3, a Component of NuRD/Mi-2 Complex, Helps Maintain Pluripotency of Mouse Embryonic Stem Cells by Repressing Trophectoderm Differentiation. PLoS ONE 2009, 4, e7684.
- Denslow, S.A.; Wade, P.A. The Human Mi-2/NuRD Complex and Gene Regulation. Oncogene 2007, 26, 5433–5438.
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a Novel Complex with Both ATP-Dependent Chromatin-Remodeling and Histone Deacetylase Activities. Mol. Cell 1998, 2, 851–861.
- Shao, S.; Cao, H.; Wang, Z.; Zhou, D.; Wu, C.; Wang, S.; Xia, D.; Zhang, D. CHD4/NuRD Complex Regulates Complement Gene Expression and Correlates with CD8 T Cell Infiltration in Human Hepatocellular Carcinoma. Clin. Epigenet. 2020, 12, 31.
- Zhang, X.; DeSalle, L.M.; Patel, J.H.; Capobianco, A.J.; Yu, D.; Thomas-Tikhonenko, A.; McMahon, S.B. Metastasis-Associated Protein 1 (MTA1) Is an Essential Downstream Effector of the c-MYC Oncoprotein. Proc. Natl. Acad. Sci. USA 2005, 102, 13968–13973.
- Fujita, N.; Jaye, D.L.; Geigerman, C.; Akyildiz, A.; Mooney, M.R.; Boss, J.M.; Wade, P.A. MTA3 and the Mi-2/NuRD Complex Regulate Cell Fate during B Lymphocyte Differentiation. Cell 2004, 119, 75–86.
- Kusam, S.; Dent, A. Common Mechanisms for the Regulation of B Cell Differentiation and Transformation by the Transcriptional Repressor Protein BCL-6. Immunol. Res. 2007, 37, 177–186.
- Morey, L.; Brenner, C.; Fazi, F.; Villa, R.; Gutierrez, A.; Buschbeck, M.; Nervi, C.; Minucci, S.; Fuks, F.; Di Croce, L. MBD3, a Component of the NuRD Complex, Facilitates Chromatin Alteration and Deposition of Epigenetic Marks. Mol. Cell. Biol. 2008, 28, 5912–5923.
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 Is a Subunit of the NuRD Complex and Targets the Metastasis Programs in Breast Cancer. Cell 2009, 138, 660–672.
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831.
- Tatetsu, H.; Kong, N.R.; Chong, G.; Amabile, G.; Tenen, D.G.; Chai, L. SALL4, the Missing Link between Stem Cells, Development and Cancer. Gene 2016, 584, 111–119.
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513.
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725.
- Smirnov, E.; Chmúrčiaková, N.; Cmarko, D. Human RDNA and Cancer. Cells 2021, 10, 3452.
- Stults, D.M.; Killen, M.W.; Williamson, E.P.; Hourigan, J.S.; Vargas, H.D.; Arnold, S.M.; Moscow, J.A.; Pierce, A.J. Human RRNA Gene Clusters Are Recombinational Hotspots in Cancer. Cancer Res. 2009, 69, 9096–9104.
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of P53. Cancer Cell 2012, 22, 51–65.