Mitochondria are central hubs in cellular physiology integrating cellular metabolism, bioenergetics, the execution of cell death and signaling through different effectors like Ca2+, reactive oxygen species (mtROS), mtDNA and different metabolites. Studies have summarized the different functions that the mitochondrial ATP synthase and its inhibitor protein, Inhibitory Factor 1 (IF1), play in cellular biology and in cancer progression. It has overviewed the mechanism by which the ATP synthase/IF1 axis contributes to metabolic reprogramming to an enhanced glycolytic phenotype, both in cancer cells and in the maintenance of stemness, and its potential both as biomarkers of prognosis and as targets for therapy. Moreover, it have highlighted how the ATP synthase/IF1 axis contributes to the signaling of cell-type specific programs that allow the adaptation of the cell/organisms to different changing cues, and finally, how the ATP synthase/IF1 axis also participates in preventing the execution of cell death and hence, in therapeutic resistance of the carcinomas. It has emphasized that the relative low activity of mitochondrial metabolic pathways, such as oxidation of pyruvate coupled to oxidative phosphorylation (OXPHOS) and β-oxidation in lung adenocarcinomas, contribute to cancer progression.
- cancer
- OXPHOS
- Warburg effect
- mitochondrial ATP synthase
- ATPase inhibitory factor 1
- metabolic reprogramming
- metastasis
- RNA binding proteins
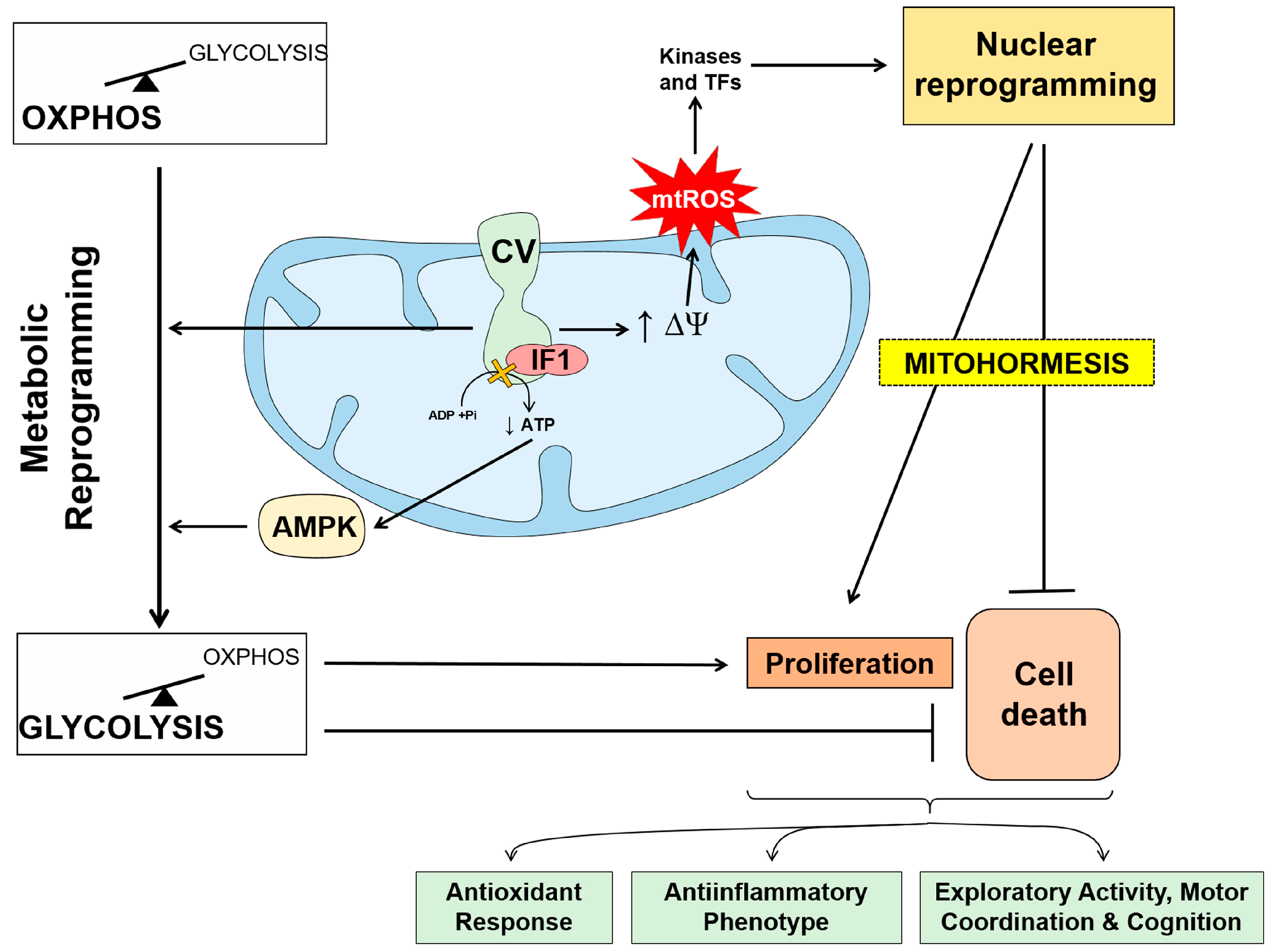
- mitohormesis
- cell death
1. Metabolic Rewiring and Cancer Progression
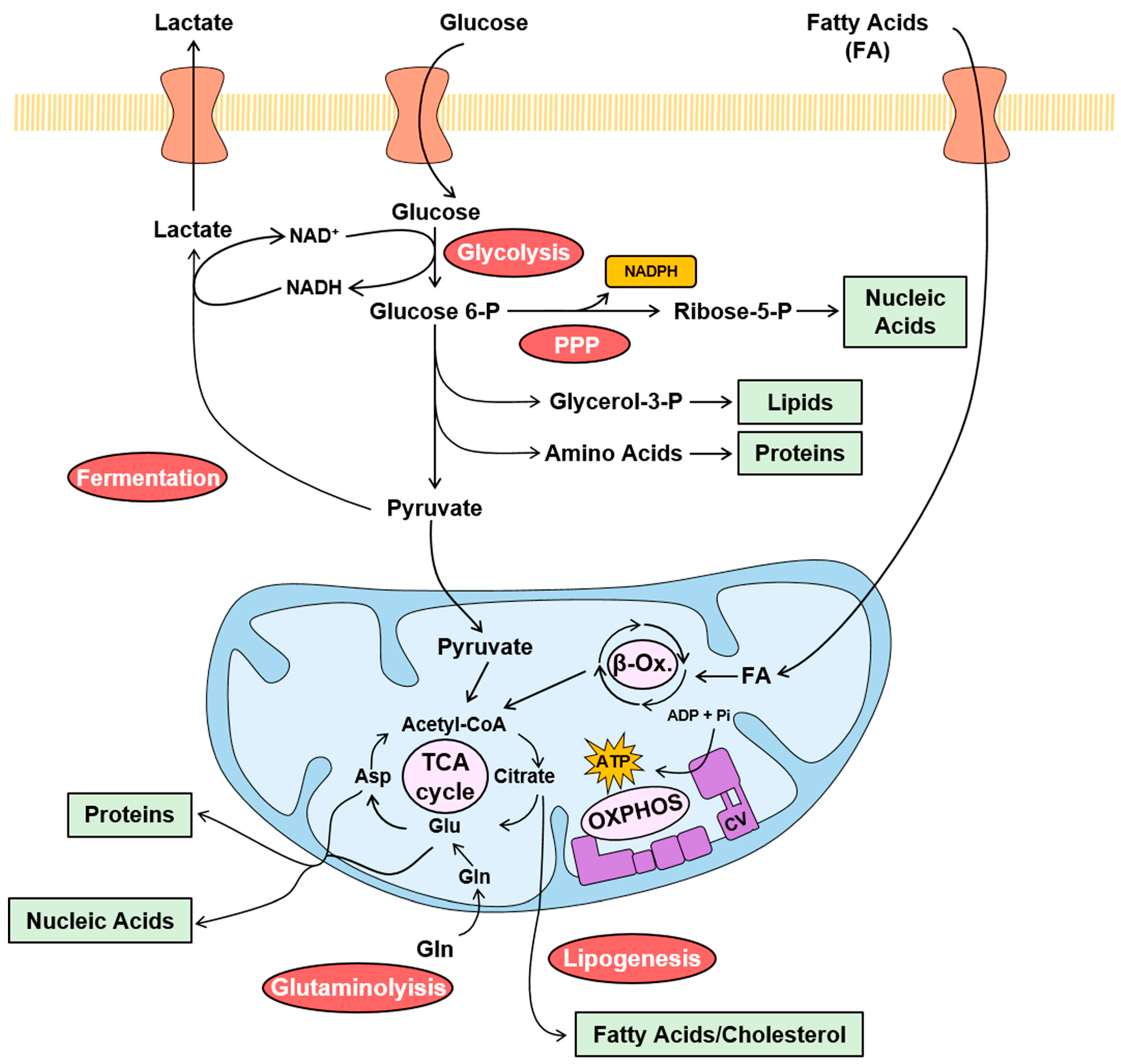
2. The Mitochondrial ATP Synthase/IF1 Axis
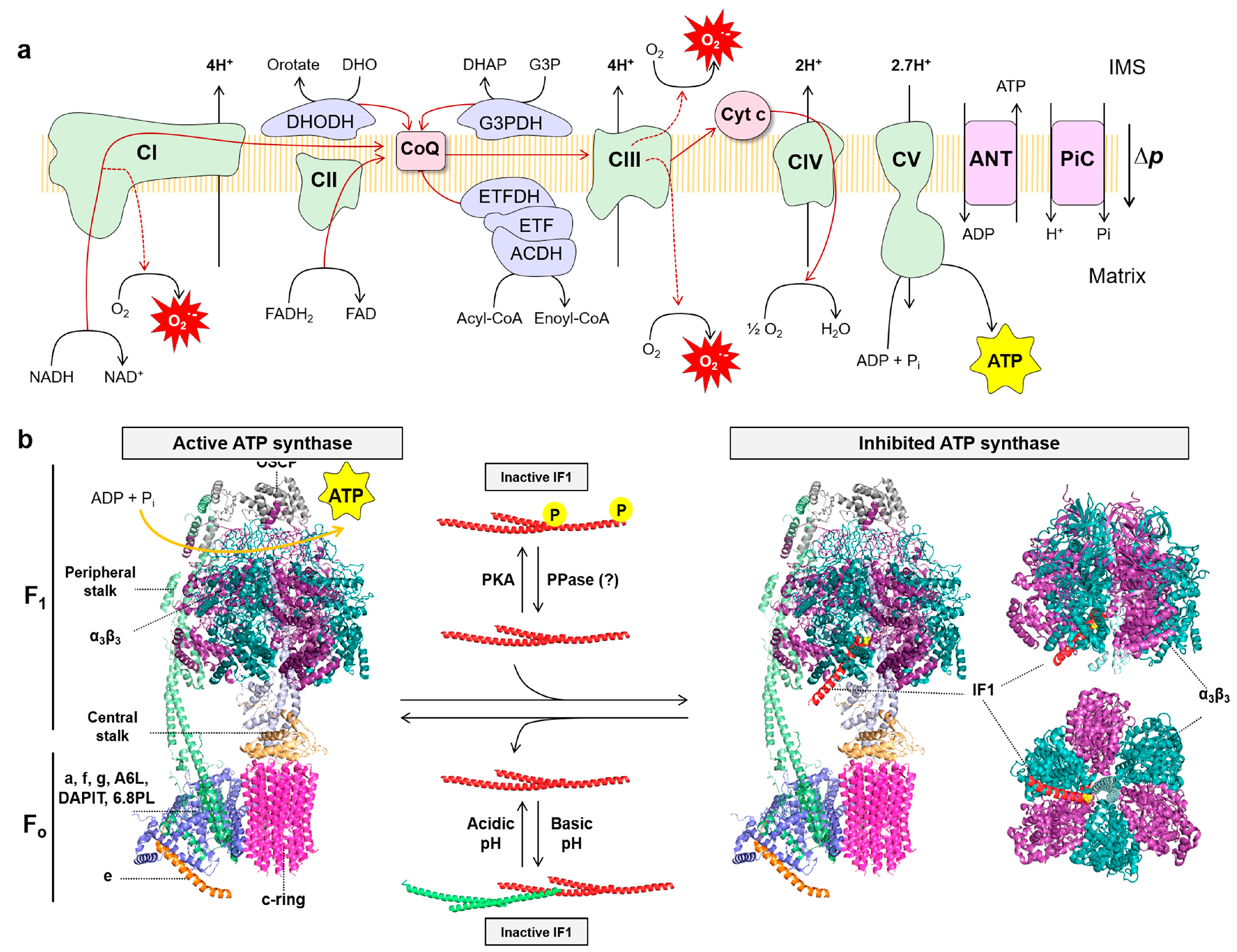

3. ATP Synthase and the Bioenergetic Signature of Cancer
5. ATPase Inhibitory Factor 1, the Physiological Inhibitor of the ATP Synthase
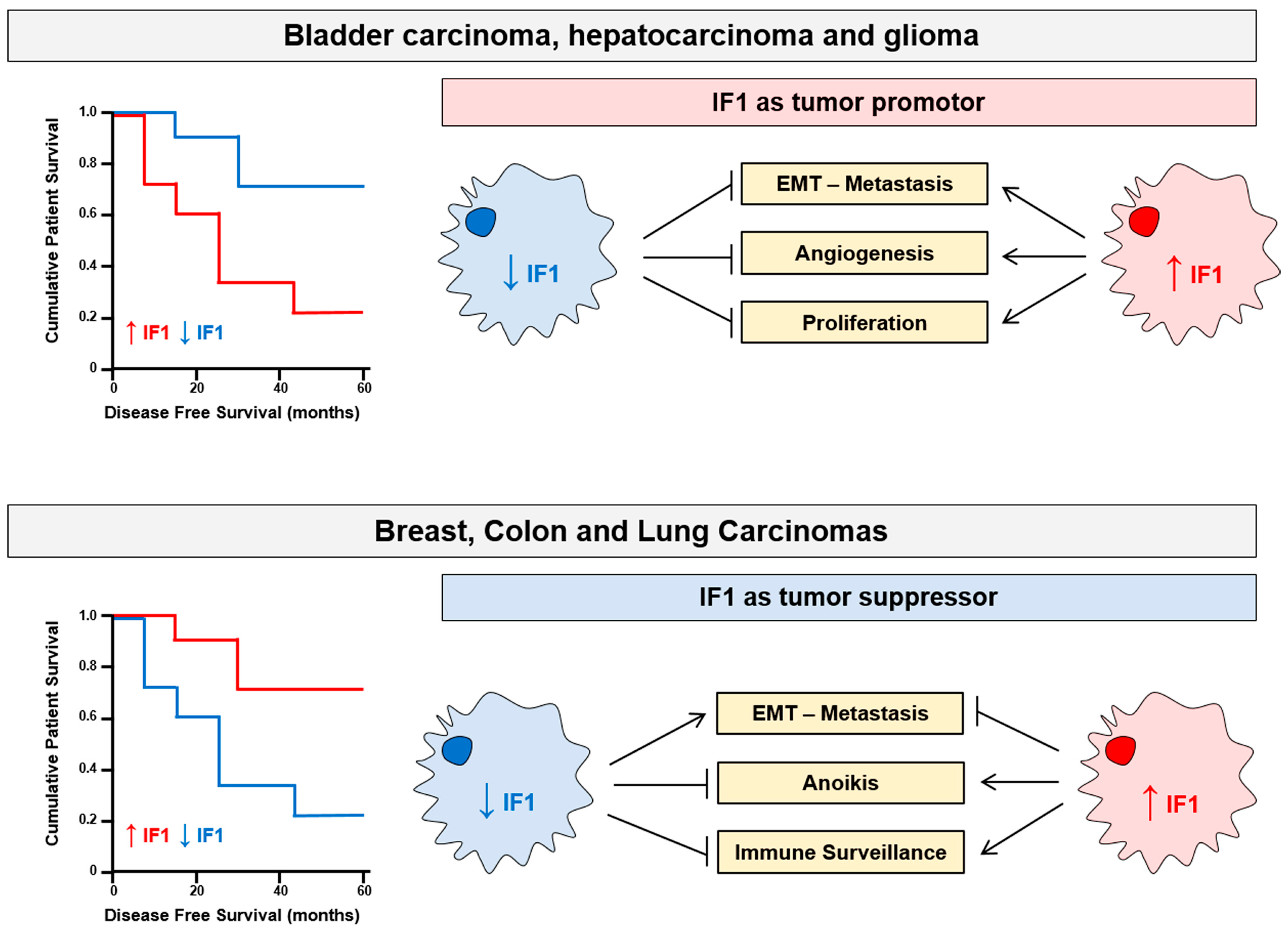
6. Tissue-Specific Activity of IF1: Tumor Promotor and Tumor Suppressor
7. Mitochondria as s Promising Target for Cancer Treatment
Novel therapeutic approaches based on personalized medicine are required to minimize the social and economic burden caused by cancer. The enzymes that control metabolism are promising targets to combat progression of the disease [3][121][122][123][124]. In this regard, several glycolytic inhibitors have been proposed and some of them are actually in clinical trials [3][69][124][125][126][127][128]. Mitochondrial metabolism also offers other effective targets to restrain tumor growth [124][127][129][130][131][132]. In this regard, antibiotics such as doxycycline that target the mitochondrial ribosome and inhibit translation prevent mitochondrial biogenesis which is required for the clonal expansion and survival of cancer stem cells (CSCs) [133]. Doxycycline has also been used in combination with other anticancer therapies [134][135][136][137] and their effectiveness lies in their ability to prevent metastasis by inhibiting the growth of CSCs.
In this line, it recently found that nebivolol, a third-generation β1-blocker, halts colon and breast tumor growth in vivo by a severe inhibition of OXPHOS [138]. Mechanistically, nebivolol binds to β1-adrenergic receptors in cancer cells, causing the increase in IF1 content bound to the ATP synthase and promoting its inhibition. Concurrently, nebivolol prevents the phosphorylation of NDUFS7, a Complex I subunit, which promotes partial inhibition of the activity of the enzyme [138]. In addition, nebivolol also arrests proliferation of endothelial cells by blocking β1-adrenergic receptors impeding tumor angiogenesis [138].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15153775
References
- Wang, R.; Green, D.R. Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 2012, 249, 14–26.
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352.
- Cuezva, J.M.; Ortega, A.D.; Willers, I.; Sanchez-Cenizo, L.; Aldea, M.; Sanchez-Arago, M. The tumor suppressor function of mitochondria: Translation into the clinics. Biochim. Biophys. Acta 2009, 1792, 1145–1158.
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2013, 76, 325–334.
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat Metab. 2020, 2, 127–129.
- Esparza-Molto, P.B.; Cuezva, J.M. Reprogramming oxidative phosphorylation in cancer: A role for RNA binding proteins. Antioxid. Redox Signal. 2020, 33, 927–945.
- Paul, S.; Ghosh, S.; Kumar, S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin. Cancer Biol. 2022, 86, 1216–1230.
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714.
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715.
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354.
- Torresano, L.; Nuevo-Tapioles, C.; Santacatterina, F.; Cuezva, J.M. Metabolic reprogramming and disease progression in cancer patients. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165721.
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544.
- Zhao, Z.; Lu, J.; Han, L.; Wang, X.; Man, Q.; Liu, S. Prognostic significance of two lipid metabolism enzymes, HADHA and ACAT2, in clear cell renal cell carcinoma. Tumour Biol. 2016, 37, 8121–8130.
- Wang, J.; Liu, F.; Ao, P.; Li, X.; Zheng, H.; Wu, D.; Zhang, N.; She, J.; Yuan, J.; Wu, X. Correlation of PDK1 expression with clinicopathologic features and prognosis of hepatocellular carcinoma. Onco Targets Ther. 2016, 9, 5597–5602.
- Lin, P.C.; Lin, J.K.; Yang, S.H.; Wang, H.S.; Li, A.F.; Chang, S.C. Expression of beta-F1-ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: Clinical data analysis and evidence from an in vitro study. Int. J. Color. Dis. 2008, 23, 1223–1232.
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144.
- Jin, L.; Chun, J.; Pan, C.; Kumar, A.; Zhang, G.; Ha, Y.; Li, D.; Alesi, G.N.; Kang, Y.; Zhou, L.; et al. The PLAG1-GDH1 Axis Promotes Anoikis Resistance and Tumor Metastasis through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol. Cell 2018, 69, 87–99.e7.
- Li, K.; Zhang, C.; Chen, L.; Wang, P.; Fang, Y.; Zhu, J.; Chen, S.; Du, J.; Shen, B.; Wu, K.; et al. The role of acetyl-coA carboxylase2 in head and neck squamous cell carcinoma. PeerJ 2019, 7, e7037.
- Osugi, J.; Yamaura, T.; Muto, S.; Okabe, N.; Matsumura, Y.; Hoshino, M.; Higuchi, M.; Suzuki, H.; Gotoh, M. Prognostic impact of the combination of glucose transporter 1 and ATP citrate lyase in node-negative patients with non-small lung cancer. Lung Cancer 2015, 88, 310–318.
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumour Biol. 2017, 39, 1010428317698338.
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391.
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273.
- Wittig, I.; Schagger, H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim. Biophys. Acta 2009, 1787, 672–680.
- Boyer, P.D. The ATP synthase. A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749.
- Kuhlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549.
- Pullman, M.E.; Monroy, G.C. A Naturally Occurring Inhibitor of Mitochondrial Adenosine Triphosphatase. J. Biol. Chem. 1963, 238, 3762–3769.
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16.
- Garcia-Bermudez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta 2016, 1857, 1167–1182.
- Sanchez-Cenizo, L.; Formentini, L.; Aldea, M.; Ortega, A.D.; Garcia-Huerta, P.; Sanchez-Arago, M.; Cuezva, J.M. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J. Biol. Chem. 2010, 285, 25308–25313.
- Formentini, L.; Sánchez-Aragó, M.; Sánchez-Cenizo, L.; Cuezva, J.M. The mitochondrial ATPase Inhibitory Factor 1 (IF1) triggers a ROS-mediated retrograde pro-survival and proliferative response. Mol. Cell 2012, 45, 731–742.
- Zhou, B.; Caudal, A.; Tang, X.; Chavez, J.D.; McMillen, T.S.; Keller, A.; Villet, O.; Zhao, M.; Liu, Y.; Ritterhoff, J.; et al. Upregulation of mitochondrial ATPase inhibitory factor 1 (ATPIF1) mediates increased glycolysis in mouse hearts. J. Clin. Investig. 2022, 132, e155333.
- Sanchez-Arago, M.; Formentini, L.; Martinez-Reyes, I.; Garcia-Bermudez, J.; Santacatterina, F.; Sanchez-Cenizo, L.; Willers, I.M.; Aldea, M.; Najera, L.; Juarranz, A.; et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis 2013, 2, e46.
- Esparza-Molto, P.B.; Romero-Carraminana, I.; Nunez de Arenas, C.; Pereira, M.P.; Blanco, N.; Pardo, B.; Bates, G.R.; Sanchez-Castillo, C.; Artuch, R.; Murphy, M.P.; et al. Generation of mitochondrial reactive oxygen species is controlled by ATPase inhibitory factor 1 and regulates cognition. PLoS Biol. 2021, 19, e3001252.
- Formentini, L.; Pereira, M.P.; Sanchez-Cenizo, L.; Santacatterina, F.; Lucas, J.J.; Navarro, C.; Martinez-Serrano, A.; Cuezva, J.M. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J. 2014, 33, 762–778.
- Santacatterina, F.; Sanchez-Cenizo, L.; Formentini, L.; Mobasher, M.A.; Casas, E.; Rueda, C.B.; Martinez-Reyes, I.; Nunez de Arenas, C.; Garcia-Bermudez, J.; Zapata, J.M.; et al. Down-regulation of oxidative phosphorylation in the liver by expression of the ATPase inhibitory factor 1 induces a tumor-promoter metabolic state. Oncotarget 2016, 7, 490–508.
- Formentini, L.; Santacatterina, F.; Nunez de Arenas, C.; Stamatakis, K.; Lopez-Martinez, D.; Logan, A.; Fresno, M.; Smits, R.; Murphy, M.P.; Cuezva, J.M. Mitochondrial ROS Production Protects the Intestine from Inflammation through Functional M2 Macrophage Polarization. Cell Rep. 2017, 19, 1202–1213.
- Esparza-Molto, P.B.; Nuevo-Tapioles, C.; Cuezva, J.M. Regulation of the H(+)-ATP synthase by IF1: A role in mitohormesis. Cell. Mol. Life Sci. 2017, 74, 2151–2166.
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206.
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569.
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766.
- Esparza-Molto, P.B.; Nuevo-Tapioles, C.; Chamorro, M.; Najera, L.; Torresano, L.; Santacatterina, F.; Cuezva, J.M. Tissue-specific expression and post-transcriptional regulation of the ATPase inhibitory factor 1 (IF1) in human and mouse tissues. FASEB J. 2019, 33, 1836–1851.
- Song, R.; Song, H.; Liang, Y.; Yin, D.; Zhang, H.; Zheng, T.; Wang, J.; Lu, Z.; Song, X.; Pei, T.; et al. Reciprocal activation between ATPase inhibitory factor 1 and NF-kappaB drives hepatocellular carcinoma angiogenesis and metastasis. Hepatology 2014, 60, 1659–1673.
- Sanchez-Gonzalez, C.; Nuevo-Tapioles, C.; Herrero Martin, J.C.; Pereira, M.P.; Serrano Sanz, S.; Ramirez de Molina, A.; Cuezva, J.M.; Formentini, L. Dysfunctional oxidative phosphorylation shunts branched-chain amino acid catabolism onto lipogenesis in skeletal muscle. EMBO J. 2020, 39, e103812.
- Dominguez-Zorita, S.; Romero-Carraminana, I.; Santacatterina, F.; Esparza-Molto, P.B.; Simo, C.; Del-Arco, A.; Nunez de Arenas, C.; Saiz, J.; Barbas, C.; Cuezva, J.M. IF1 ablation prevents ATP synthase oligomerization, enhances mitochondrial ATP turnover and promotes an adenosine-mediated pro-inflammatory phenotype. Cell Death Dis. 2023, 14, 413.
- Gu, J.; Zhang, L.; Zong, S.; Guo, R.; Liu, T.; Yi, J.; Wang, P.; Zhuo, W.; Yang, M. Cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science 2019, 364, 1068–1075.
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085.
- Gross, A.; Pilcher, K.; Blachly-Dyson, E.; Basso, E.; Jockel, J.; Bassik, M.C.; Korsmeyer, S.J.; Forte, M. Biochemical and genetic analysis of the mitochondrial response of yeast to BAX and BCL-X(L). Mol. Cell. Biol. 2000, 20, 3125–3136.
- Santamaria, G.; Martinez-Diez, M.; Fabregat, I.; Cuezva, J.M. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H+-ATP synthase. Carcinogenesis 2006, 27, 925–935.
- Dey, R.; Moraes, C.T. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J. Biol. Chem. 2000, 275, 7087–7094.
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J.C. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1998, 1, 327–336.
- Mnatsakanyan, N.; Park, H.A.; Wu, J.; He, X.; Llaguno, M.C.; Latta, M.; Miranda, P.; Murtishi, B.; Graham, M.; Weber, J.; et al. Mitochondrial ATP synthase c-subunit leak channel triggers cell death upon loss of its F1 subcomplex. Cell Death Differ. 2022, 29, 1874–1887.
- Bernardi, P.; Carraro, M.; Lippe, G. The mitochondrial permeability transition: Recent progress and open questions. FEBS J. 2021, 289, 7051–7074.
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285.
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459.
- Kinnally, K.W.; Campo, M.L.; Tedeschi, H. Mitochondrial channel activity studied by patch-clamping mitoplasts. J. Bioenerg. Biomembr. 1989, 21, 497–506.
- Petronilli, V.; Szabo, I.; Zoratti, M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 1989, 259, 137–143.
- Gerle, C. Mitochondrial F-ATP synthase as the permeability transition pore. Pharmacol. Res. 2020, 160, 105081.
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662.
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360.
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892.
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585.
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683.
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat Commun. 2019, 10, 5823.
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414.
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Torresano, L.; Santacatterina, F.; Dominguez-Zorita, S.; Nuevo-Tapioles, C.; Nunez-Salgado, A.; Esparza-Molto, P.B.; Gonzalez-Llorente, L.; Romero-Carraminana, I.; Nunez de Arenas, C.; Sanchez-Garrido, B.; et al. Analysis of the metabolic proteome of lung adenocarcinomas by reverse-phase protein arrays (RPPA) emphasizes mitochondria as targets for therapy. Oncogenesis 2022, 11, 24.
- Sanchez-Arago, M.; Chamorro, M.; Cuezva, J.M. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010, 31, 567–576.
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681.
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409.
- D’Errico, I.; Salvatore, L.; Murzilli, S.; Lo Sasso, G.; Latorre, D.; Martelli, N.; Egorova, A.V.; Polishuck, R.; Madeyski-Bengtson, K.; Lelliott, C.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. USA 2011, 108, 6603–6608.
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481.
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127.
- Liu, W.; Beck, B.H.; Vaidya, K.S.; Nash, K.T.; Feeley, K.P.; Ballinger, S.W.; Pounds, K.M.; Denning, W.L.; Diers, A.R.; Landar, A.; et al. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014, 74, 954–963.
- Ikari, R.; Mukaisho, K.I.; Kageyama, S.; Nagasawa, M.; Kubota, S.; Nakayama, T.; Murakami, S.; Taniura, N.; Tanaka, H.; Kushima, R.P.; et al. Differences in the Central Energy Metabolism of Cancer Cells between Conventional 2D and Novel 3D Culture Systems. Int. J. Mol. Sci. 2021, 22, 1805.
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640.
- Fernandez-Vizarra, E.; Enriquez, J.A.; Perez-Martos, A.; Montoya, J.; Fernandez-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213.
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123.
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446.
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.C.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734.
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694.
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384.
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793.
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301.
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001-1015.
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632.
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605.
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209.
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev. 2018, 32, 230–243.
- Isidoro, A.; Martinez, M.; Fernandez, P.L.; Ortega, A.D.; Santamaria, G.; Chamorro, M.; Reed, J.C.; Cuezva, J.M. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem. J. 2004, 378, 17–20.
- Lopez-Rios, F.; Sanchez-Arago, M.; Garcia-Garcia, E.; Ortega, A.D.; Berrendero, J.R.; Pozo-Rodriguez, F.; Lopez-Encuentra, A.; Ballestin, C.; Cuezva, J.M. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007, 67, 9013–9017.
- Xu, J.Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e17.
- Cuezva, J.M.; Chen, G.; Alonso, A.M.; Isidoro, A.; Misek, D.E.; Hanash, S.M.; Beer, D.G. The bioenergetic signature of lung adenocarcinomas is a molecular marker of cancer diagnosis and prognosis. Carcinogenesis 2004, 25, 1157–1163.
- Ricart, J.; Egea, G.; Izquierdo, J.M.; San Martin, C.; Cuezva, J.M. Subcellular structure containing mRNA for beta subunit of mitochondrial H+-ATP synthase in rat hepatocytes is translationally active. Biochem. J. 1997, 324 Pt 2, 635–643.
- Lithgow, T.; Cuezva, J.M.; Silver, P.A. Highways for protein delivery to the mitochondria. Trends Biochem. Sci. 1997, 22, 110–113.
- Egea, G.; Izquierdo, J.M.; Ricart, J.; San Martín, C.; Cuezva, J.M. mRNA encoding the beta-subunit of the mitochondrial F1-ATPase complex is a localized mRNA in rat hepatocytes. Biochem. J. 1997, 322, 557–565.
- Izquierdo, J.M.; Cuezva, J.M. Control of the translational efficiency of beta-F1-ATPase mRNA depends on the regulation of a protein that binds the 3’ untranslated region of the mRNA. Mol. Cell. Biol. 1997, 17, 5255–5568.
- Willers, I.M.; Isidoro, A.; Ortega, A.D.; Fernandez, P.L.; Cuezva, J.M. Selective inhibition of beta-F1-ATPase mRNA translation in human tumours. Biochem. J. 2010, 426, 319–326.
- Izquierdo, J.M.; Cuezva, J.M. Internal-ribosome-entry-site functional activity of the 3’-untranslated region of the mRNA for the beta subunit of mitochondrial H+-ATP synthase. Biochem. J. 2000, 346, 849–855.
- Di Liegro, C.M.; Bellafiore, M.; Izquierdo, J.M.; Rantanen, A.; Cuezva, J.M. 3’-untranslated regions of oxidative phosphorylation mRNAs function in vivo as enhancers of translation. Biochem. J. 2000, 352 Pt 1, 109–115.
- Ortega, A.D.; Willers, I.M.; Sala, S.; Cuezva, J.M. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J. Cell Sci. 2010, 123, 2685–2696.
- Willers, I.M.; Cuezva, J.M. Post-transcriptional regulation of the mitochondrial H(+)-ATP synthase: A key regulator of the metabolic phenotype in cancer. Biochim. Biophys. Acta 2011, 1807, 543–551.
- Cabezon, E.; Butler, P.J.; Runswick, M.J.; Walker, J.E. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J. Biol. Chem. 2000, 275, 25460–25464.
- Cabezon, E.; Butler, P.J.; Runswick, M.J.; Carbajo, R.J.; Walker, J.E. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J. Biol. Chem. 2002, 277, 41334–41341.
- Sanchez-Arago, M.; Formentini, L.; Garcia-Bermudez, J.; Cuezva, J.M. IF1 reprograms energy metabolism and signals the oncogenic phenotype in cancer. Cell Cycle 2012, 11, 2963–2964.
- Kahancova, A.; Sklenar, F.; Jezek, P.; Dlaskova, A. Regulation of glucose-stimulated insulin secretion by ATPase Inhibitory Factor 1 (IF1). FEBS Lett. 2018, 592, 999–1009.
- Kahancova, A.; Sklenar, F.; Jezek, P.; Dlaskova, A. Overexpression of native IF1 downregulates glucose-stimulated insulin secretion by pancreatic INS-1E cells. Sci. Rep. 2020, 10, 1551.
- Garcia-Bermudez, J.; Sanchez-Arago, M.; Soldevilla, B.; Del Arco, A.; Nuevo-Tapioles, C.; Cuezva, J.M. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H(+)-ATP Synthase. Cell Rep. 2015, 12, 2143–2155.
- Gonzalez-Llorente, L.; Santacatterina, F.; Garcia-Aguilar, A.; Nuevo-Tapioles, C.; Gonzalez-Garcia, S.; Tirpakova, Z.; Toribio, M.L.; Cuezva, J.M. Overexpression of Mitochondrial IF1 Prevents Metastatic Disease of Colorectal Cancer by Enhancing Anoikis and Tumor Infiltration of NK Cells. Cancers 2019, 12, 22.
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544.
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968.
- Sanchez-Arago, M.; Garcia-Bermudez, J.; Martinez-Reyes, I.; Santacatterina, F.; Cuezva, J.M. Degradation of IF1 controls energy metabolism during osteogenic differentiation of stem cells. EMBO Rep. 2013, 14, 638–644.
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594.
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271.
- Christensen, G.L.; Kelstrup, C.D.; Lyngso, C.; Sarwar, U.; Bogebo, R.; Sheikh, S.P.; Gammeltoft, S.; Olsen, J.V.; Hansen, J.L. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol. Cell. Proteom. 2010, 9, 1540–1553.
- Garcia-Aguilar, A.; Martinez-Reyes, I.; Cuezva, J.M. Changes in the turnover of the cellular proteome during metabolic reprogramming: A role for mtROS in proteostasis. J. Proteome Res. 2019, 18, 3142–3155.
- Shen, L.; Zhi, L.; Hu, W.; Wu, M.X. IEX-1 targets mitochondrial F1Fo-ATPase inhibitor for degradation. Cell Death Differ. 2009, 16, 603–612.
- Wei, S.; Fukuhara, H.; Kawada, C.; Kurabayashi, A.; Furihata, M.; Ogura, S.; Inoue, K.; Shuin, T. Silencing of ATPase Inhibitory Factor 1 Inhibits Cell Growth via Cell Cycle Arrest in Bladder Cancer. Pathobiology 2015, 82, 224–232.
- Wu, J.; Shan, Q.; Li, P.; Wu, Y.; Xie, J.; Wang, X. ATPase inhibitory factor 1 is a potential prognostic marker for the migration and invasion of glioma. Oncol. Lett. 2015, 10, 2075–2080.
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15.
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31.
- Sanchez-Arago, M.; Cuezva, J.M. The bioenergetic signature of isogenic colon cancer cells predicts the cell death response to treatment with 3-bromopyruvate, iodoacetate or 5-fluorouracil. J. Transl. Med. 2011, 9, 19.
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649.
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180.
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551.
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242.
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346.
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584.
- Lamb, R.; Fiorillo, M.; Chadwick, A.; Ozsvari, B.; Reeves, K.J.; Smith, D.L.; Clarke, R.B.; Howell, S.J.; Cappello, A.R.; Martinez-Outschoorn, U.E.; et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget 2015, 6, 14005–14025.
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with Doxycycline: Implications for optimizing anti-angiogenic therapy. Oncotarget 2017, 8, 56126–56142.
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269–67286.
- Ozsvari, B.; Magalhaes, L.G.; Latimer, J.; Kangasmetsa, J.; Sotgia, F.; Lisanti, M.P. A Myristoyl Amide Derivative of Doxycycline Potently Targets Cancer Stem Cells (CSCs) and Prevents Spontaneous Metastasis, without Retaining Antibiotic Activity. Front. Oncol. 2020, 10, 1528.
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Nunez de Arenas, C.; Gomez de Cedron, M.; Formentini, L.; Cuezva, J.M. Coordinate beta-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat Commun. 2020, 11, 3606.