Mitochondria are central hubs in cellular physiology integrating cellular metabolism, bioenergetics, the execution of cell death and signaling through different effectors like Ca2+, reactive oxygen species (mtROS), mtDNA and different metabolites. Studies have summarized the different functions that the mitochondrial ATP synthase and its inhibitor protein, IF1, play in cellular biology and in cancer progression. It has overviewed the mechanism by which the ATP synthase/IF1 axis contributes to metabolic reprogramming to an enhanced glycolytic phenotype, both in cancer cells and in the maintenance of stemness, and its potential both as biomarkers of prognosis and as targets for therapy. Moreover, it have highlighted how the ATP synthase/IF1 axis contributes to the signaling of cell-type specific programs that allow the adaptation of the cell/organisms to different changing cues, and finally, how the ATP synthase/IF1 axis also participates in preventing the execution of cell death and hence, in therapeutic resistance of the carcinomas. It has emphasized that the relative low activity of mitochondrial metabolic pathways, such as OXPHOS and β-oxidation in lung adenocarcinomas, contribute to cancer progression.
1. Metabolic Rewiring and Cancer Progression
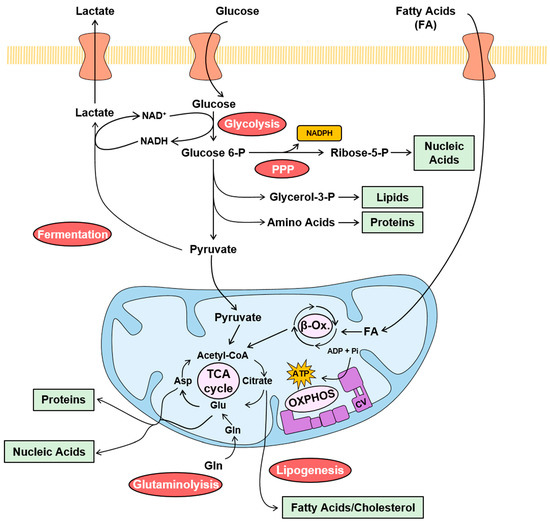
Carcinomas undergo a coordinated reprogramming of their metabolic pathways to satisfy the demand for high energy and for the building blocks that are required to sustain cellular proliferation [
21,
22,
26,
27,
28,
29]. The metabolic reprogramming experienced by cancer is generally known as the Warburg effect, and it is characterized by the sharp increase in the rates of glycolysis and of the fermentation of pyruvate into lactate, which allows the regeneration of NAD
+ for glycolysis to proceed at high rates (
Figure 1). Glycolysis and fermentation do not require oxygen to function. Concurrent with the hyperactivation of glycolysis and fermentation, the oxidation of pyruvate coupled to oxidative phosphorylation (OXPHOS) in mitochondria, which is strictly dependent on the availability of oxygen, is marginally increased and/or partially inhibited in cancer cells, to spare the backbone of carbon skeletons for the synthesis of precursors (
Figure 1) [
26,
29,
30,
31]. In fact, the citrate formed in the TCA cycle is exported to the cytoplasm for providing the acetyl-CoA that is used for the synthesis of fatty acids and cholesterol (
Figure 1) [
32]. However, cancer cells replenish carbon skeletons in the TCA cycle in the form of glutamate by increasing the metabolism of glutamine (
Figure 1) [
33]. Simultaneously, the metabolism of glucose through the pentose phosphate pathway (PPP) is also significantly augmented, to produce the ribose that is used for building nucleotides and producing the NADPH that is used in biosynthetic processes and in the maintenance of the cellular redox state (
Figure 1) [
34]. Consistent with the reprogramming of metabolism in carcinomas, the sharp increase in different enzymes of glycolysis provide biomarkers that predict a bad patient prognosis [
35], whereas, for example, the silencing of glycolytic enzymes in glioblastoma (GBM) xenografts dramatically increase the survival of mice [
36]. In contrast, the loss of mitochondrial proteins involved in pyruvate oxidation, β-oxidation and OXPHOS predicts bad patient prognosis [
35,
37,
38,
39], supporting the theory that a diminished metabolic activity of the organelle compromises survival. Likewise, the overexpression of enzymes of metabolic pathways that are induced in carcinomas, such as glutamine metabolism [
35,
40,
41] and the biosynthesis of fatty acids [
35,
42,
43,
44,
45], predicts bad patient prognosis.
Figure 1. Metabolic pathways in cancer. The color-coded scheme illustrates the major cancer-promoted changes in relevant metabolic pathways observed in human carcinomas. Glycolysis, fermentation, pentose phosphate pathway (PPP), lipogenesis and glutaminolysis are enhanced (red circles) in carcinomas. However, the mitochondrial oxidation of pyruvate, β-oxidation (β-Ox.), tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) are marginally increased (pink circles) or diminished when compared to the activation of metabolic pathway in the cytoplasm. Glucose enters the cell by specific transporters and is metabolized at high rate through glycolysis to provide precursors such as glycerol-3-P and amino acids for lipid and protein synthesis (green box), respectively. Pyruvate is reduced to lactate in fermentation to regenerate NAD+ to allow glycolysis to proceed and is exported from the cell. In addition, glucose is catabolized through the PPP to obtain NADPH (orange box), for biosynthetic processes and the maintenance of the cellular redox state, and ribose-5-P, for nucleic acid biosynthesis (green box). In the mitochondria, pyruvate is oxidized to generate acetyl-CoA that feeds the TCA cycle. Fatty acids (FA) oxidation also feeds acetyl-CoA to the TCA cycle. Citrate in the TCA cycle is drained to the cytoplasm for the biosynthesis of FA and cholesterol. Glutaminolysis feeds the TCA cycle with glutamate. The electrons obtained in the oxidation of pyruvate and FA are transferred to the electron transport chain system for the synthesis of the ATP by OXPHOS (purple).
2. The Mitochondrial ATP Synthase/IF1 Axis
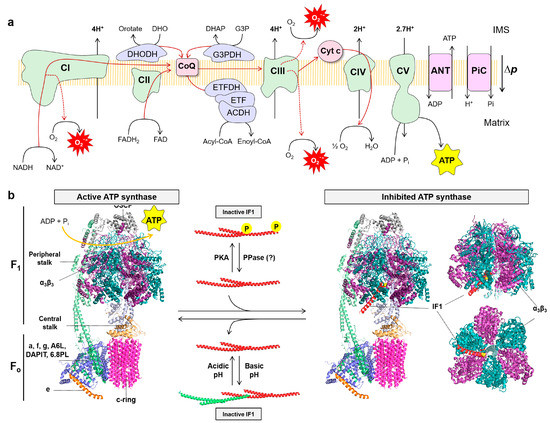
The OXPHOS system integrates the complexes of the electron transport chain (ETC) (complex I–IV), a variety of different dehydrogenases, the ATP synthase, two diffusible electron carriers such as CoQ and cytochrome c, and substrate transporters in the inner mitochondrial membrane (IMM) (
Figure 2a) [
30,
67,
68]. Respiratory complexes of the ETC, and a variety of FAD-dependent dehydrogenases that feed electrons directly to CoQ, transfer electrons obtained in biological oxidations in the form of NADH and FADH
2 down to molecular oxygen, to generate the proton electrochemical gradient responsible for the proton-motive force (∆
p) across the IMM (
Figure 2a). The ATP synthase utilizes ∆
p by coupling the backflow of protons into the matrix of the organelle for the generation of ATP from ADP and inorganic phosphate (Pi) (
Figure 2a).
Figure 2. The mitochondrial OXPHOS system: (a) The OXPHOS system consists of five protein complexes: complexes I–IV (CI–IV) of the electron transport chain (ETC) and complex V (CV) or ATP synthase. In addition, there are mobile electron carriers such as coenzyme Q (CoQ) and cytochrome c (cyt c), as well as membrane transporters that include among others the pyruvate carrier (not shown), adenine nucleotide translocase (ANT) and the phosphate carrier (PiC). Other FAD-dependent dehydrogenases transfer electrons to CoQ, such as glycerol-3-phosphate dehydrogenase (G3PDH), dihydroorotate dehydrogenase (DHOH), acyl-CoA dehydrogenase complex (ACDH), electron transfer flavoprotein (ETF) and electron transfer flavoprotein dehydrogenase (ETFDH). The electron transfer from NADH and FADH2 to molecular oxygen (O2), the final electron acceptor, builds a proton-motive force (∆p) by the pumping of protons through complexes I, III and IV, which is used by ATP synthase to produce ATP; (b) Structure of monomeric bovine active ATP synthase (left) and bound to the N-terminal inhibitory fragment of IF1 in the inhibited ATP synthase state (right). The soluble F1 domain is composed of α3β3 subunits (purple/blue) and the central stalk (γ subunit, light blue, and δ, ε subunits, light yellow), while the Fo domain is formed by a ring of 8 c subunits (pink) and a subunit (light blue). These two domains are linked by a peripheral stalk, composed of b, d, F6 subunits (light green), and the oligomycin sensitivity-conferring protein (OSCP; grey). The supernumerary subunits include e (orange), f, g, A6L, the protein associated with diabetes in insulin-sensitive tissues (DAPIT), and the 6.8 kDa proteolipid (6.8PL) (light blue). Right, the interaction between the N-terminal inhibitory fragment of IF1 (red) and the α3β3 and γ subunits is shown. The Ala14 residue of IF1 (Ser14 in human and mouse IF1) is highlighted in yellow. The active ATP synthase is able to synthetize ATP using ADP and Pi. When IF1 is bound it is inhibited and no longer synthetizes nor hydrolyzes ATP. Regulation of the ATP synthase is exerted by IF1. IF1 can be inactivated by forming oligomers that mask the inhibitory regions that bind to the F1 domain when the matrix pH is above neutrality. However, in mitochondrial de-energization conditions the pH drops below neutrality and IF1 becomes activated. On the other hand, IF1 can be phosphorylated in S39 by PKA preventing its interaction with the ATP synthase. IF1 has a short-half life and there is no evidence yet for the existence of an IF1 protein phosphatase (PPase). Molecular reconstruction from PDB: 6ZPO, 1OHH and 1GMJ. Images created using the PyMOL molecular graphics system.
The ATP synthase, a protein complex composed of 18 different proteins in humans, drives the production of ATP using the free energy stored in the proton-motive force (
Figure 2a,b) [
69]. It consists of two main domains, the F
o embedded in the IMM and the F
1 domain projected into the matrix, containing the
α and
β subunits of the catalytic core (
Figure 2b) [
70]. The F
o and F
1 domains are linked together by a central (subunit
γ,
ε and
δ) and a peripheral (subunit
b,
d, OSCP and F6) stalk (
Figure 2b). The protons are imported into the matrix through subunit a causing the rotation of the
c-ring in the F
o domain. The central stalk transfers this rotational energy to the
α3β3 subunits in the F
1 domain, leading to the synthesis of ATP (
Figure 2b).
The ATP synthase has a small inhibitory protein, the ATPase Inhibitory Factor 1 (IF1), that for long time has been considered an inhibitor only (unidirectional) of the hydrolytic activity of the enzyme by binding the
αβ interface in the F
1 domain (
Figure 2b) [
71,
72]. IF1 is a highly conserved protein among mammalian species and is encoded in the nuclear
ATP5IF1 gene [
73].
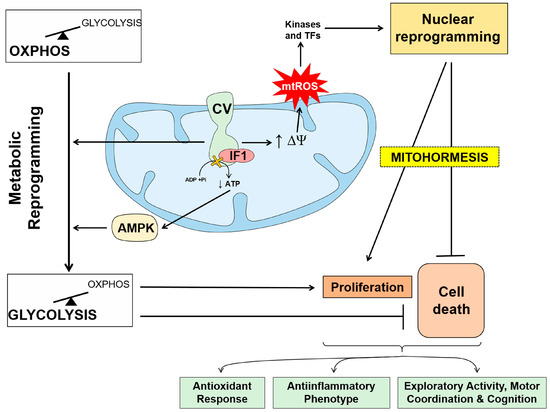
The IF1-mediated inhibition of ATP synthase results in partial blockade of the import of protons into mitochondria, the limitation of OXPHOS activity and the metabolic reprogramming of the cells to an enhanced glycolysis (
Figure 3) [
74,
75,
84,
85]. Moreover, the blockage in proton import also promotes mitochondrial hyperpolarization and the subsequent production of ROS (mtROS) at respiratory complexes of the ETC (
Figure 3) [
75,
82]. The generated mtROS modify the activity of protein kinases and of transcription factors that signal to the nucleus different programs that allow adaptation of the cell to changing cues (
Figure 3) [
75,
79,
80,
81,
82]. In some mouse tissues, the IF1-mediated activation of mtROS signaling promotes long-lasting metabolic and molecular cytoprotective mechanisms that allow cells to withstand subsequent insults (
Figure 3) [
79,
80,
81], a process known as mitohormesis [
1,
2,
3,
86]. However, in other mouse tissues which are naturally devoid of IF1 [
87], the overexpression of IF1 is detrimental [
80,
88,
89]. In sharp contrast, the overexpression of IF1 in neurons protects from excitotoxic insults [
79] and increases the exploratory activity, motor coordination and cognition of mice (
Figure 3) [
82]. Similarly, the overexpression of IF1 in intestinal epithelial cells provides protection against inflammation (
Figure 3) [
81], whereas IF1 ablation results in a pro-inflammatory phenotype of fatal consequences [
83]. Overall, these results emphasized the relevance of the ATP synthase/IF1 axis in signaling and controlling tissue-specific and non-cell autonomous cell fate decisions.
Figure 3. The role of ATP synthase/IF1 axis in metabolic reprogramming and signaling. Binding of IF1 to the ATP synthase limits the production of ATP and promotes metabolic reprogramming towards an enhanced glycolytic phenotype by allosteric regulation of the enzymes of glycolysis and the activation of AMPK. Inhibition of ATP synthase prevents the backflow of protons into the matrix increasing the mitochondrial membrane potential (∆Ψm). The increase in ∆Ψm leads to an increase in the generation of mitochondrial reactive oxygen species (mtROS) at the electron transport chain that signal to the nucleus through kinases and transcription factors. Nuclear reprogramming promotes the activation of cellular responses that include proliferation, prevention of cell death allowing adaptation of the cell to changing cues in mitohormetic responses. The cellular signaling response mediated by IF1 overexpression is cell type specific in mouse tissues and promotes antioxidant responses, anti-inflammatory phenotypes and motor coordination and cognition. In contrast, IF1 overexpression in skeletal muscle and heart is deleterious.
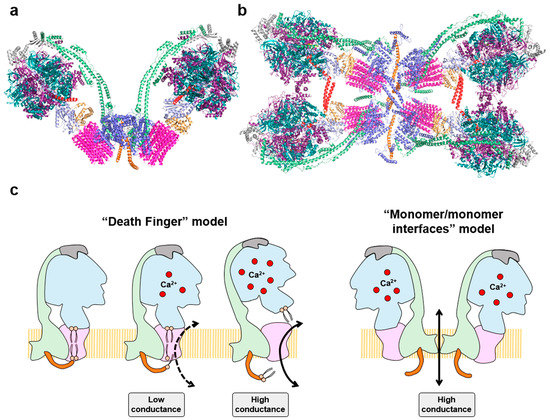
Besides its function in energy provision and in signaling, the ATP synthase also plays a structural role in the IMM by forming dimers and oligomers to shape cristae at its rims (
Figure 4a,b) [
70]. Recent cryo-EM structures of mammalian tetrameric ATP synthases (
Figure 4b) [
90,
91] and isobaric quantitative Protein Interaction Reporter (iqPIR) cross-linking technologies [
84], have uncovered the structural role that IF1 plays in oligomerization and inhibition of ATP synthase, and in reprogramming of the tissue to an enhanced glycolysis [
84].
Figure 4. Oligomeric assemblies of the ATP synthase and the permeability transition pore (PTP): (a) Structure of bovine dimeric ATP synthase promotes the curvature of the inner mitochondrial membrane at its tips. Molecular reconstruction from PDB: 7AJD; (b) Cryo-electron microscopy structure of porcine IF1-inhibited ATP synthase tetramers viewed from the matrix side. The IF1 dimers (red) bind to two adjacent antiparallel ATP synthase dimers. Molecular reconstruction from PDB: 6J5K. Images created using the PyMOL molecular graphics system. Both in a, and b, the color coding of the subunits is the same as in Figure 2; (c) Two major models explain the participation of the ATP synthase in the PTP. The “death finger” model proposes that Ca2+ binding to β subunit causes a rearrangement in the F1 domain increasing its rigidity that is transmitted through OSCP (grey) to the peripheral stalk (green) and to the e subunit (orange). Subunit e exerts a pulling force on the outer lipids that plug the center of the c-ring, leading to the formation of a channel that allows the entrance of ions. Mild Ca2+ levels lead to closed and open states in a low-conductance mode of opening (“flickering”) in the PTP. However, when Ca2+ increases the channel is more prone to opening, promoting complete displacement of inner lipids in the c-rotor and of the central stalk of the ATP synthase, committing cells to death by the high-conductance PTP. In the “monomer/monomer interfaces” model, the Ca2+ induced conformational changes in dimers of the ATP synthase destabilize the dimeric assembly opening a high conductance pore in the interface of the dimer (subunits e and g).
Moreover, the ATP synthase is also a critical component in the regulation and execution of cell death since its components [
92] and activity [
93,
94,
95] are required for the efficient execution of cell death. In fact, the ATP synthase is structurally and functionally implicated in permeability transition (mPT) [
96,
97,
98]. mPT is exerted through a high-conductance channel in the IMM, known as the Permeability Transition Pore (PTP) [
99,
100,
101]. PTP opening leads to mitochondrial swelling and cell death and is regulated by Ca
2+ and/or cyclophilin D-dependent structural changes on ATP synthase [
97,
102], whereas it is inhibited by cyclosporine A (CsA) [
103,
104]. The ATP synthase is postulated as a key component of the PTP [
96,
97,
105,
106,
107,
108], although its participation in mPT has been questioned [
109,
110]. Nowadays, the participation of the ATP synthase in PTP is undisputed after the reunification of the different alternatives that implicated the enzyme in the “death finger” and “monomer/monomer interfaces” models of the PTP (
Figure 4c) [
96,
97,
98,
102].
3. ATP Synthase and the Bioenergetic Signature of Cancer
As previously mentioned, the high rates of pyruvate fermentation observed in cancer cells under aerobic conditions lead Warburg to suggest that carcinomas had an impaired bioenergetic activity of mitochondria [
114]. However, while the upregulation of glycolysis in carcinomas is nowadays out of the question, the alteration of mitochondrial bioenergetics is still a matter of debate, despite the obvious evidence linking the down-regulation of mitochondrial metabolism and bioenergetics in cancer progression [
20,
35,
51,
115,
116,
117,
118,
119,
120]. Perhaps the debate emerges due to the metabolic behavior of some cancer cells when growing in culture [
121], differences in the mitochondrial proteome and activity in different tissues [
122,
123], the heterogeneity of the microenvironment of carcinomas [
124,
125,
126,
127,
128] and/or the finding that metastatic disease requires an enhanced expression of OXPHOS proteins [
129,
130,
131,
132,
133,
134,
135].
Indeed, the down-regulation of the catalytic subunit of mitochondrial ATP synthase (β-F1-ATPase) is observed in a large number of human carcinomas [
20,
35,
115,
140], either when expressed in absolute levels or normalized relative to other mitochondrial proteins, such as Hsp60 and/or to the glycolytic GAPDH. In fact, the quotient of the expression level of these three biomarkers β-F1-ATPase/Hsp60/GAPDH offers a bioenergetic cellular (BEC) index that informs about the overall capacity of mitochondria in the tissue or carcinoma [
26,
115].
Remarkably, a high-throughput quantitative analysis of 27 biomarkers of metabolism—using Reverse Phase Protein Array (RPPA) technology, in a cohort of 128 tumors and the corresponding paired NAT of patients bearing lung adenocarcinomas (LUAD)— confirmed that the BEC index is significantly diminished in the carcinomas, suggesting that OXPHOS activity is limited in LUAD [
20], in agreement with previous reports [
16,
23,
145].
The downregulation of the mitochondrial ATP synthase in cancer can be mediated at the expression and activity levels [
30]. The expression of the catalytic subunit of ATP synthase (β-F1-ATPase) in mammalian liver is complex, including the subcellular localization of its mRNA in a structure that is frequently found attached to the outer mitochondrial membrane [
156,
157]. β-F1-ATPase mRNA localization and translation requires at least two cis-acting elements and a large set of RNABPs [
156,
158] and the 3′-UTR of the mRNA that acts as a translational enhancer sequence [
159,
160,
161,
162].
An affinity purification approach of RNABPs using as bait the full-length β-F1-ATPase mRNA allowed the identification of nine bona fide mRNA binding proteins [
170]. Among them, G3BP1 (Ras-GTPase-Activating Protein SH3-Domain-Binding Protein) was found to interact both in vivo and in vitro with the 3′UTR of β-F1-ATPase mRNA, to promote inhibition of the synthesis of the protein by preventing mRNA translation [
30,
166,
170].
5. ATPase Inhibitory Factor 1, the Physiological Inhibitor of the ATP Synthase
As mentioned above, the control of the overall ATP synthase activity is not only exerted at the protein level, but also by regulation of the content and activity of IF1, its inhibitor protein. For many years, it has been considered that under normal physiological situations, i.e., when the mitochondrial matrix pH is above neutrality, IF1 forms inactive oligomers by masking the inhibitory regions that bind to the F
1 domain of the enzyme (
Figure 2b) [
175,
176]. Only when mitochondria become de-energized, and matrix pH drops below neutrality, IF1 is considered to become activated by depolymerization and binds to the ATP synthase to inhibit its hydrolase activity (
Figure 2b) [
175,
176].
Studies in metabolic reprograming in human carcinomas revealed that normal lung, colon and breast tissues express negligible quantities of IF1 [
85] but showed a sharp increase in the expression of the protein in the carcinomas and in cultured cancer cells [
74,
75,
85]. These findings raised the reasonable hypothesis that IF1 might represent a sort of mitochondrial “oncogene” [
178], not only to control the hydrolytic activity of the ATP synthase, as it was postulated many years ago, but also its ATP synthetic activity. In other words, the overexpression of IF1 could be mediating the partial inhibition of OXPHOS contributing to the metabolic reprogramming experienced in oncogenesis. Indeed, the overexpression of IF1 in several cancer cells [
74,
75,
77,
78,
85], mouse models of gain of function of IF1 [
79,
80,
81,
82,
84,
89] and pharmacologic in vivo approaches [
76], amply demonstrated that IF1 overexpression inhibits the ATP synthetic activity of ATP synthase in isolated mitochondria, diminished tissue ATP levels, activated AMPK and promoted the metabolic rewiring of cells and tissues to an enhanced glycolytic phenotype, i.e., induced the Warburg phenotype. Conversely, silencing or knocking out IF1 in cancer cells [
20,
179] and mouse models of loss of function of IF1 [
82,
83] resulted in and enhanced activity of the ATP synthase.
Stem cells, iPSC and cancer cells have a predominant glycolytic phenotype whereas OXPHOS prevails in differentiated cells [
180,
181]. Interestingly, IF1 also plays a relevant role in metabolic reprogramming during differentiation of human mesenchymal stem cells (hMSC) and in the maintenance/acquisition of stemness [
182]. In fact, hMSC also present high levels of IF1, altered mitochondrial structure and molecular composition, low rates of OXPHOS and an enhanced glycolytic metabolism [
182].
Remarkably, the results in tissues of mouse models with regulated expression of IF1 further suggest that mitochondria in non-stressed physiological conditions of some cellular types such as cardiomyocytes [
76], neurons [
82] and intestinal epithelial cells [
83] contain a fraction of IF1-bound and -inhibited ATP synthase, in agreement with recent cryo-EM studies of mammalian heart ATP synthases [
90,
91].
The regulation of IF1 activity as an inhibitor of ATP synthase is also exerted by covalent modification of the protein by phosphorylation (
Figure 2b). Phosphoproteomic studies indicated that serine residues of IF1 are phosphorylated in cancer cells [
185,
186,
187].
Indeed, the phosphorylation of IF1 has been observed when there is an increase in energy demand in the heart in vivo or in cells in cultures when they are forced to increase OXPHOS [
76]. Likewise, IF1 is phosphorylated when cells progress through the OXPHOS-dependent G1-phase of the cell cycle [
76]. In contrast, hypoxia and progression through the reductive phases of the cell cycle (S/G2/M) promote dephosphorylation of IF1, the reduction of OXPHOS and an enhanced glycolytic flux [
76]. Consistently, dephosphorylated IF1 is the prevalent form of the inhibitor protein in lung, colon, and breast carcinomas, contributing to the inhibition of ATP synthase and the reprogramming of metabolism to an enhanced glycolysis and fermentation [
76].
Most available evidence suggests that regulation of IF1 expression is exerted at post-transcriptional levels by controlling the rates of its synthesis and degradation [
182]. In fact, the increase in protein content of IF1 in carcinomas [
85] or its sharp reduction during cellular differentiation [
182], are exerted in the absence of relevant changes in the abundance of IF1-mRNA. In fact, the IF1 protein has a very high turnover rate (2–3 h), both in cancer cells [
85] and in differentiated osteocytes [
182], which is much faster than the turnover of many other subunits of ATP synthase in cancer cells (18 h) [
193]. IF1 degradation is mediated by several mitochondrial serine proteases [
85,
182] and metalloproteases [
194], but the specific genes involved have not been identified yet [
182].
6. Tissue-Specific Activity of IF1: Tumor Promotor and Tumor Suppressor
A fundamental characteristic of IF1 is that it is a tissue-specific and species-specific expressed protein in mouse and human tissues [
87]. Human normal tissues such as the heart, brain, kidney, stomach, endometrium and liver have a high IF1 content, whereas breast, colon, and lung epithelia contain negligible levels of IF1 [
85,
87]. Likewise, in human carcinomas the expression of IF1 is also variable [
85]. Whereas oncogenesis in epithelial cells of the endometrium, stomach and kidney does not promote an increase in IF1 expression, carcinomas in the colon, lung and breast show a very sharp increase in the content of IF1 [
85].
In agreement with the pro-oncogenic role of IF1, studies in human hepatocellular carcinomas (HCC) showed that high levels of IF1 predict worse overall and progression free survival for the patients (
Figure 5) [
88]. Mechanistically, IF1 activates the non-canonical NFκB pathway, driving angiogenesis and metastasis through the activation of SNAI1 and VEGF [
88]. Consistent with these findings, transgenic mice overexpressing in the liver the constitutively active mutant IF1-H49K promoted the inhibition of OXPHOS and a state of metabolic preconditioning guided by the activation of the stress kinases AMPK and p38 MAPK [
80]. Diethylnitrosamine-induced liver carcinogenesis in IF1-H49K transgenic mice contributed to an enhanced liver carcinogenesis by augmenting proliferation and apoptotic resistance of carcinoma cells [
80]. Likewise, the overexpression of IF1 in bladder carcinomas [
190] and in gliomas [
191] also predict a worse prognosis for the patients and a shorter time to disease recurrence (
Figure 5). Mechanistically, the silencing of IF1 diminished the rates of proliferation, migration and invasive capacities of the cells, preventing epithelial mesenchymal transition (EMT) (
Figure 5) [
88,
190,
191]. In contrast, the overexpression of IF1 in these cancer cells promoted angiogenesis and activation of EMT, increasing the expression of E-cadherin and diminishing that of vimentin, to promote their metastatic capacity (
Figure 5) [
88,
190,
191].
7. Mitochondria as s Promising Target for Cancer Treatment
Novel therapeutic approaches based on personalized medicine are required to minimize the social and economic burden caused by cancer. The enzymes that control metabolism are promising targets to combat progression of the disease [9,14,26,200,201]. In this regard, several glycolytic inhibitors have been proposed and some of them are actually in clinical trials [26,51,152,201,202,203,204]. Mitochondrial metabolism also offers other effective targets to restrain tumor growth [201,203,205,206,207,208]. In this regard, antibiotics such as doxycycline that target the mitochondrial ribosome and inhibit translation prevent mitochondrial biogenesis which is required for the clonal expansion and survival of cancer stem cells (CSCs) [209]. Doxycycline has also been used in combination with other anticancer therapies [210,211,212,213] and their effectiveness lies in their ability to prevent metastasis by inhibiting the growth of CSCs.
In this line, it recently found that nebivolol, a third-generation β1-blocker, halts colon and breast tumor growth in vivo by a severe inhibition of OXPHOS [60]. Mechanistically, nebivolol binds to β1-adrenergic receptors in cancer cells, causing the increase in IF1 content bound to the ATP synthase and promoting its inhibition. Concurrently, nebivolol prevents the phosphorylation of NDUFS7, a Complex I subunit, which promotes partial inhibition of the activity of the enzyme [60]. In addition, nebivolol also arrests proliferation of endothelial cells by blocking β1-adrenergic receptors impeding tumor angiogenesis [60].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15153775