Targeted therapies for solid tumors, including non-small cell lung cancer (NSCLC), have advanced significantly, offering tailored treatment options for patients. However, individuals without targetable mutations pose a clinical challenge, as they may not respond to standard treatments like immune-checkpoint inhibitors (ICIs) and novel targeted therapies; further, there is an unmet need to identify prognostic biomarkers of response to treatment. Inflammatory cytokines such as interleukin-1 beta (IL-1β) have emerged as a key area of focus and hold significant potential implications for future clinical practice. Combinatorial approaches of IL-1β inhibitors and ICIs may provide a potential therapeutic modality for NSCLC patients without targetable mutations and offer insight into the continued search for prognostic biomarkers.
- ICIs
- IL-1β
- therapeutic resistance
- inflammation
- signaling
- NSCLC
- tumor microenvironment
1. Introduction
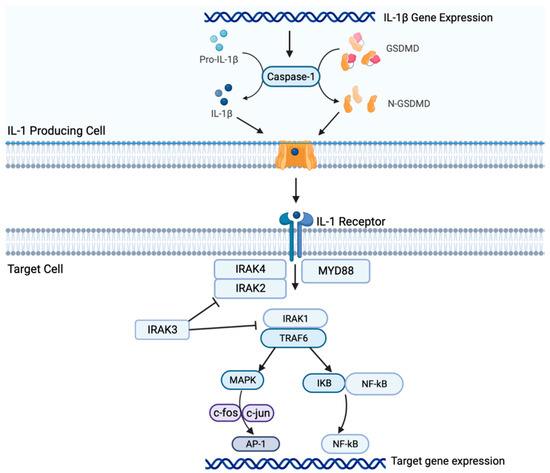
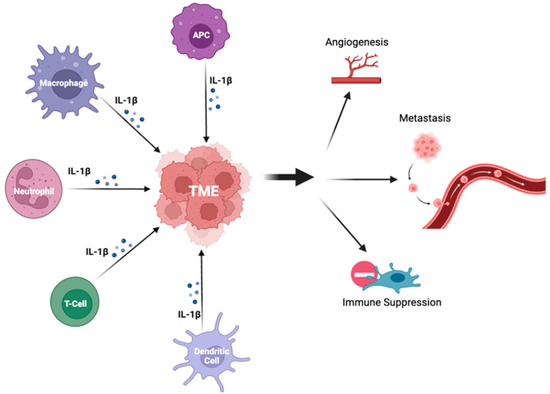
2. Il-1β Signaling in NSCLC
3. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/ijms241411547
References
- Viviane Teixeira L. de Alencar; Amanda B. Figueiredo; Marcelo Corassa; Kenneth J. Gollob; Vladmir C. Cordeiro de Lima; Lung cancer in never smokers: Tumor immunology and challenges for immunotherapy. Front. Immunol. 2022, 13, 984349, .
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R.; Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009, .
- Ming Yi; Mengke Niu; Linping Xu; Suxia Luo; Kongming Wu; Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10, .
- Roy S. Herbst; Daniel Morgensztern; Chris Boshoff; The biology and management of non-small cell lung cancer. Nat. 2018, 553, 446-454, .
- Xi Yan; Lina Han; Riyang Zhao; Sumaya Fatima; Lianmei Zhao; Feng Gao; Prognosis value of IL-6, IL-8, and IL-1β in serum of patients with lung cancer: A fresh look at interleukins as a biomarker. Heliyon 2022, 8, e09953, .
- Cédric Rébé; François Ghiringhelli; Interleukin-1β and Cancer. Cancers 2020, 12, 1791, .
- Shuhang Wang; Pei Yuan; Beibei Mao; Ning Li; Jianming Ying; Xiuli Tao; Wei Tang; Lei Zhang; Xiao Geng; Fan Zhang; et al. Genomic features and tumor immune microenvironment alteration in NSCLC treated with neoadjuvant PD-1 blockade. npj Precis. Oncol. 2022, 6, 1-8, .
- Jun Zhang; Nirmal Veeramachaneni; Targeting interleukin-1β and inflammation in lung cancer. Biomark. Res. 2022, 10, 1-9, .
- Xiang Qin; Tingting Li; Shun Li; Hong Yang; Chunhui Wu; Chuan Zheng; Fengming You; Yiyao Liu; The tumor biochemical and biophysical microenvironments synergistically contribute to cancer cell malignancy. null 2019, 17, 1186-1187, .
- Jay M Lee; Masahiro Tsuboi; Edward S Kim; Tony Sk Mok; Pilar Garrido; Overcoming immunosuppression and pro-tumor inflammation in lung cancer with combined IL-1β and PD-1 inhibition. Futur. Oncol. 2022, 18, 3085-3100, .
- Moshe Elkabets; Vera S. G. Ribeiro; Charles A. Dinarello; Suzanne Ostrand-Rosenberg; James P. Di Santo; Ron N. Apte; Christian A. J. Vosshenrich; IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347-3357, .
- Shengjun Li; Wei Wang; Ning Zhang; Tingxian Ma; Chenghai Zhao; IL-1β mediates MCP-1 induction by Wnt5a in gastric cancer cells. BMC Cancer 2014, 14, 480-480, .
- Ryan Kolb; Liem Phan; Nicholas Borcherding; Yinghong Liu; Fang Yuan; Ann M. Janowski; Qing Xie; Kathleen R. Markan; Wei Li; Matthew J. Potthoff; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007, .
- Yaron Carmi; Shahar Dotan; Peleg Rider; Irena Kaplanov; Malka R. White; Rona Baron; Shai Abutbul; Monica Huszar; Charles A. Dinarello; Ron N. Apte; et al. The Role of IL-1β in the Early Tumor Cell–Induced Angiogenic Response. null 2013, 190, 3500-3509, .
- Jian Huang; Xiuwen Lan; Ting Wang; Hailing Lu; Mengru Cao; Shi Yan; Yue Cui; Dexin Jia; Li Cai; Ying Xing; et al. Targeting the IL-1β/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene 2019, 39, 1739-1755, .
- Erin Fahey; Sarah L. Doyle; IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426, .
- Pei Hai Ping; Tan Feng Bo; Liu Li; Yu Nan Hui; Zhu Hong; IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch. Biochem. Biophys. 2016, 604, 20-26, .
- Chen Wu; Bin Xu; You Zhou; Mei Ji; Dachuan Zhang; Jingting Jiang; Changping Wu; Correlation between serum IL-1β and miR-144-3p as well as their prognostic values in LUAD and LUSC patients. Oncotarget 2016, 7, 85876-85887, .
- M. F. Sanmamed; J. L. Perez-Gracia; K. A. Schalper; J. P. Fusco; A. Gonzalez; M. E. Rodriguez-Ruiz; C. Oñate; G. Perez; C. Alfaro; S. Martín-Algarra; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988-1995, .
- Seth B. Coffelt; Kelly Kersten; Chris W. Doornebal; Jorieke Weiden; Kim Vrijland; Cheei-Sing Hau; Niels J. M. Verstegen; Metamia Ciampricotti; Lukas J. A. C. Hawinkels; Jos Jonkers; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nat. 2015, 522, 345-348, .
- Chengcheng Jin; Georgia K. Lagoudas; Chen Zhao; Susan Bullman; Arjun Bhutkar; Bo Hu; Samuel Ameh; Demi Sandel; Xu Sue Liang; Sarah Mazzilli; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998-1013.e16, .
- Nicholas L Syn; Michele W L Teng; Tony S K Mok; Ross A Soo; De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731-e741, .
- Edward B. Garon; James Chih-Hsin Yang; Steven M. Dubinett; The Role of Interleukin 1β in the Pathogenesis of Lung Cancer. JTO Clin. Res. Rep. 2020, 1, 100001, .
- Valerio Gelfo; Donatella Romaniello; Martina Mazzeschi; Michela Sgarzi; Giada Grilli; Alessandra Morselli; Beatrice Manzan; Karim Rihawi; Mattia Lauriola; Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009, .
- Ningning Yan; Sanxing Guo; Huixian Zhang; Ziheng Zhang; Shujing Shen; Xingya Li; BRAF-Mutated Non-Small Cell Lung Cancer: Current Treatment Status and Future Perspective. Front. Oncol. 2022, 12, 863043, .
- Eva Hajek; Franziska Krebs; Rebekka Bent; Katharina Haas; Antje Bast; Ivo Steinmetz; Andrea Tuettenberg; Stephan Grabbe; Matthias Bros; BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, 9, 28294-28308, .
- Angela M. Davies; Primo N. Lara; Philip C. Mack; Paul H. Gumerlock; Richard J. Bold; David R. Gandara; Bortezomib-Based Combinations in the Treatment of Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2005, 7, S59-S63, .
- Allyson G. McLoed; Taylor P. Sherrill; Dong-Sheng Cheng; Wei Han; Jamie A. Saxon; Linda A. Gleaves; Pingsheng Wu; Vasiliy V. Polosukhin; Michael Karin; Fiona E. Yull; et al. Neutrophil-Derived IL-1β Impairs the Efficacy of NF-κB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120-132, .
- Yuan, B.; Clowers, M.J.; Velasco, W.V.; Peng, S.; Peng, Q.; Shi, Y.; Ramos-Castaneda, M.; Zarghooni, M.; Yang, S.; Babcock, R.L.; et al. Targeting IL-1β as an immunopreventive and therapeutic modality for K-ras-mutant lung cancer.. JCI Insight 2022, 7, e157788, .