The drug metabolism and drug degradation pathways may overlap, resulting in the formation of similar constituents. Therefore, the metabolism data can be helpful for deriving safe levels of degradation impurities and improving the quality of respective pharmaceutical products. The present article contains considerations on possible links between metabolic and degradation pathways for new antidiabetic drugs such as glutides, gliflozins, and gliptins. Special attention was paid to their reported metabolites and identified degradation products. At the same time, many interesting analytical approaches to conducting metabolism as well as degradation experiments were mentioned, including chromatographic methods and radioactive labeling of the drugs.
1. Introduction
Type 2 diabetes mellitus (T2DM) is one of the most common metabolic diseases. It results from decreased secretion by pancreatic b-cells and insulin resistance. In turn, alterations in insulin acting in peripheral tissues lead to elevated blood glucose levels with severe metabolic disturbances, e.g., macro- and micro-vascular complications. Thus, the treatment of hyperglycemia should target not only impaired insulin secretion but also hepatic and muscular insulin resistance, reduced intestinal incretin effects, and increased glucose renal threshold. Optimal management of T2DM should also consider other risk factors beyond glycemic control, i.e., excess body weight, cardiovascular risk, heart failure, and renal diseases [
1,
2,
3]. Therefore, typical initial drugs to treat T2DM, i.e., insulin and sulfonylureas, are not optimal for every patient because of the associated risk of hypoglycemia and weight gain. An increase in body weight in diabetic patients potentially leads to increased insulin resistance and cardiovascular risk. Therefore, the preferred drugs are those that do not increase or even lower the body weight, such as glucagon-like peptide 1 (GLP-1) receptor agonists (GLP-1 RAs or glutides) and sodium glucose co-transporter 2 inhibitors (SGLT2 inhibitors or gliflozins). Therefore, combined treatment with a GLP-1 RA and an SGLT2 inhibitor, with or without metformin, could be the recommended initial therapy for T2DM for many patients. As a result, in addition to lowering glucose levels, a decrease in cardiac events, cardiac mortality, and the rate of kidney damage could be expected [
1,
4,
5].
Drug metabolism is a complicated process in which drugs can be modified by various endogenous enzymes. Experiments on the drug metabolism are important steps in the process of modifying new molecules for their optimal therapeutic properties, identifying new active compounds based on the structures of active metabolites, enhancing drug safety due to the elimination of toxic metabolites, and comparing the drug metabolism in animals and humans for predicting optimal human doses [
6,
7,
8]. Despite the human and animal studies, in vitro metabolism experiments, which are relatively inexpensive and easy to carry out, could be used as a reliable screening procedure to recognize the drug metabolites, which could explain the particular metabolic pathways and designate directions for further in vivo experiments. These in vitro models include the use of slices from liver and kidney tissues, recombinant human cytochromes (CYPs), other enzymes, chemical inhibitors, and co-factors. Essential points for conducting such in vitro studies include examination of appropriate concentrations of substrates, co-factors, enzymes and their inhibitors, and, finally, the drugs being tested. In addition, the important steps of experiments, such as the time of incubation and time of sampling, as well as all steps of method validation, should be determined with great care. Finally, the data from in vitro experiments should be analyzed in relation to animal and human studies to predict the consequences of drug metabolism in patients. After an incubation with enzymes and all needed ingredients, the samples from these in vitro experiments are usually analyzed with high-performance liquid chromatography combined with high-resolution mass spectrometry (HPLC-HRMS). This combination is used because of its excellent sensitivity, selectivity, accuracy, and precision of quantification. The accurate mass measurement allows identification of individual components without the need to separate them, simplifying analysis and interpretation of the results [
9]. The next step of such experiments is the investigation of the structures of the detected metabolites in relation to their parent drug, in which the main metabolic pathways of phase I, i.e., oxidation, reduction, and hydrolysis, and phase II, i.e., acetylation, sulfation, and glucuronidation, should be taken into account.
Even with the possibility of using various software and databases, the identification of unknown compounds, including metabolites or degradation products, is still a complex task. The measurement of accurate masses, the correlation of retention values from chromatography, and fragmentation spectra from MS are often difficult and may be insufficient for the complete identification of many compounds. For reliable identification of metabolites, it is usually necessary to increase the concentration of the analytes and carry out counter-synthesis. Thus, concurrent experiments with or without inserting particular isotope labels should be carried out, as it is a credible tool for the identification of these unknown molecules. The stable isotope labeling provides a huge elevation in both metabolite identification and metabolic pathway proposal. Such stable isotopes have the same number of protons as common elements, and consequently share the same physicochemical properties, but they differ in mass due to a difference in the number of neutrons. Among biochemically relevant elements, carbon, hydrogen, nitrogen, oxygen, and sulfur that have two or more stable isotopes with measurable abundance can be used in the labeling of the samples.
Chromatographic techniques still make up the majority of the methods used for the separation, isolation, and purification of drugs and their metabolites from biological samples, as well as their degradation products. However, after the separation and isolation are accomplished, specialized procedures and techniques are required for the identification of the particular products. The standard spectroscopic techniques, such as ultraviolet absorption (UV), mass spectrometry (MS), infrared spectroscopy (IR), and nuclear magnetic resonance (NMR), may be used off-line after metabolite or degradant isolation or coupled with liquid (HPLC or LC) or gas (GC) chromatographic methods. Today, the GC-MS and LC-MS methods are the prevailing analytic tools in metabolism and degradation studies. Compared with GC-MS, the advantage of LC-MS is that there is no need for chemical derivatization and, generally, faster run times for the analytes. With the introduction of ultra-performance liquid chromatography (UPLC) and highly accurate MS, the application of UPLC-MS has tremendously increased when compared with traditional methods [
6,
11].
2. Analytical Tools for Peptide Drugs Examination
The best methods for the separation of peptide products include HPLC, UPLC, ion exchange high performance liquid chromatography (HPLC-IEX), and size exclusion high-performance liquid chromatography (HPLC-SEC). Furthermore, the high sensitivity and selectivity of MS coupled with LC or UPLC (HRMS) are suitable for the identification of peptides, their metabolites, and degradation products. For peptide compounds, the most widely used ionization modes in MS are electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) techniques [
15]. Mass spectrometers can also be combined with a quadrupole, such as a Q-TOF system, in which fragmentation can increase the selectivity of these methods. Furthermore, ion trap and orbitrap mass analyzers that deliver excellent resolution and mass accuracy could be applied. These accurate HRMS systems detect a wide range of compounds as well as small molecules during both targeted and untargeted analyses without losing selectivity or sensitivity. Due to its high specificity, HRMS allows measuring the majority of peptides, including those with changed sequences, even if they are not completely separated. Even when changes to the primary amino acid sequence in peptides due to degradation or other changes occur, they could be recorded as detectable mass shifts. Furthermore, metabolomics studies of peptide drugs are frequently performed using HRMS [
11,
15].
Generally, peptides can be separated with the most frequently used C18 columns; however, some of them can give poor peak shape or peak tailing. For these reasons, C4 columns are recommended for the separation of peptides. Furthermore, phenyl columns, which are similar to C4 columns as far as their hydrophobicity is concerned, could be used for peptide analysis. In the literature, the usefulness of polar embedded or polar endcapped columns, in which polar interactions between peptides and the particle surface are enhanced, is also reported. In addition, peptide drugs could be more effectively separated using longer columns than those usually used in the separation of small drug molecules. Thus, columns with a length of 15 or 25 cm are recommended. As far as mobile phases are concerned, the reversed phase (RP) mode of chromatography could be used for peptides, but it usually requires the use of ion-pair reagents in order to achieve a good peak shape for the analytes [
16]. Among the best-rated ion pair reagents for such analysis is trifluoroacetic acid (TFA), which can be added to the mobile phase at a concentration of 0.1%. In consequence, the mobile phases used for peptide separations in RP systems and containing TFA are generally adjusted to a low pH. At such conditions, the carboxylic groups of amine acids are non-ionized and only slightly polar. On the other hand, when the pH of the mobile phase rises to 6–7, the carboxylic groups tend to ionize, making the peptide less hydrophobic.
To carry out a forced degradation study of macromolecules, generally, temperature, pH change, and agitation are used as factors to check the instability of the desired molecules. However, small peptides should be subjected to acidic, basic, and neutral hydrolysis, oxidation, photostability, and thermal treatment, according to the strategy of ICH guidelines Q1A(R2) [
17], just like the small molecule drugs. At the same time, the selection of appropriate stress conditions is essential and constitutes the most crucial step in conducting the decisive forced degradation study. It is recommended to optimize the stress conditions in such a way to attain degradation kinetics thermodynamically equivalent to accelerated stability conditions [
18]. It is known that excessive stress can lead to the secondary degradation of products.
3. Glutides (GLP-1 RAs)
To ameliorate the altered incretin effects in T2DM, glutides or the GLP-1 RAs, i.e., albiglutide (ALBI), dulaglutide (DULA), exenatide (EXE), liraglutide (LIRA), lixisenatide (LIXI) in injectable formulations, and semaglutide (SEMA) in injectable and oral formulations, are introduced as effective therapeutic approaches. It is well known that the incretin effect contributes nearly half of the insulin secretory response to oral glucose load and that this effect is reduced along with worsening glucose tolerance in diabetic patients. The glucose-dependent insulinotropic polypeptide, formerly known as gastric inhibitory peptide (GIP), and glucagon-like peptide-1 (GLP-1) are the two primary incretins secreted from the intestine on ingestion of glucose to stimulate insulin secretion from pancreatic b-cells, enhance satiety, and delay gastric emptying. GIP and GLP-1 exert their effects by binding to their specific receptors, the GIP receptor and the GLP-1 receptor (GLP-1 R), which both belong to the G-protein-coupled receptor family. Physiologically, GIP and GLP-1 are rapidly degraded by the enzyme dipeptidyl peptidase 4 (DPP-4), which acts on peptides to cleave the two NH
2-terminal amino acids [
20,
21]. Thus, GIP and GLP-1 share common properties as incretins, but they also possess different biological characteristics. Endogenous GIP exerts strong insulinotropic effects in healthy subjects, but its insulinotropic effect is seriously reduced in diabetic patients, which discourages the development of GIP-based therapies. In contrast, the insulinotropic effect of GLP-1 is preserved in T2DM patients, establishing GLP-1 and GLP-1R signaling as attractive therapeutic targets [
21]. GLP-1 RAs exert a dual action on the endocrine pancreas cells, with a stimulation of insulin secretion by b-cells, mainly in the postprandial state, and an inhibition of glucagon secretion by a-cells, contributing to reducing hyperglycemia. In addition, they slow down gastric emptying and activate the hindbrain GPL-1 receptors.
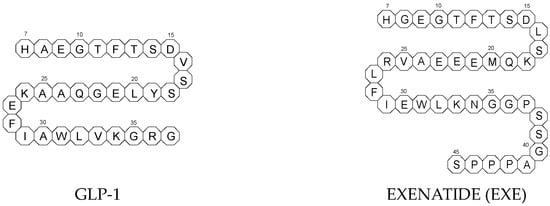
All glutides present on the market are synthetic peptides with high homology with human GLP-1 (7–37). EXE, which is given to patients subcutaneously twice daily, was the first GLP-1 RA introduced into therapy in 2005 (FDA approval). It is a 39-amino acid synthetic peptide with 53% homology with human GLP-1 (7–37). It contains an Ala8Gly substitution that increases its resistance to degradation by DPP-4.
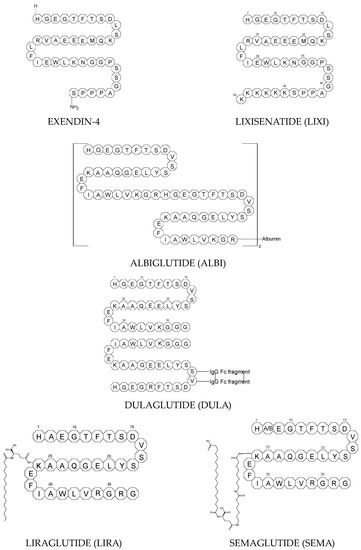
Next GLP-1 RAs, including subcutaneously once daily administered LIRA and LIXI and once weekly given ALBI and DULA, were approved by FDA in 2021, 2016, and 2014, respectively. LIRA is a true analogue of GLP-1 (97% homology) with the addition of a 16-carbon palmitic acid chain conjugated via a glutamate spacer at Lys in position 26, to mask the DPP-4 cleavage site. LIXI is an exendin-4 analogue (94% sequence homology with GLP-1) with 6 Lys residues added to allow resistance to DPP-4. ALBI consists of 2 copies of GLP-1, each with an Ala8Gly substitution, and as a whole, the molecule is fused to albumin [
23]. DULA has two copies of a GLP-1 analogue (with amino acid substitutions Ala8Gly, Gly22Glu, and Arg36Gly) that are covalently linked to an Fc fragment of human IgG4 [
24].
The next GLP-1 RA, i.e., SEMA, is marketed as a once-weekly subcutaneous injection and was approved by the FDA in 2017. It is an analogue of LIRA with a substitution of Ala at position 8 with an aminoisobutyric acid (A/b). The C16 fatty acid is also exchanged for the C18 fatty acid and linked by a synthetic spacer. Acylation with a spacer and C-18 fatty acid chain increases its binding to blood albumin, which enables its longer presence in the blood circulation. Despite the above-mentioned subcutaneous form of SEMA, its orally administered analog with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl]amino)caprylate (SNAC) to overcome the problems of poor absorption and degradation in the stomach was approved in 2019 [
21,
25]. The structures of the mentioned GLP-1 RAs are presented in
Figure 1.
Figure 1. Chemical structures of GLP-1 RAs agonists: albiglutide (ALBI), dulaglutide (DULA), GLP-1, exenatide (EXE), exendin-4, liraglutide (LIRA), lixisenatide (LIXI) and semaglutide (SEMA).
3.1. Metabolic Transformations of Glutides (GLP-1 RAs) and the Methods Used to Examine Their Metabolic Pathways
The HRMS method was used to assess the metabolism of different GLP-1 RAs, i.e., exenatide, lixisenatide, liraglutide, and semaglutide (EXE, LIXI, LIRA, and SEMA) in subcutaneous tissue of rats, minipigs, and humans [
26]. For the separation of these drugs and their metabolites, two types of columns were used, depending on the analyzed peptide, i.e., XSelect CSH C18 XP and UPLC Peptide CSH C18 (Waters, Milford, MA, USA). Analyses were conducted with an orbitrap mass spectrometer operating in ESI-positive full scan/data-dependent tandem MS (MS/MS). It was shown that LIXI was completely metabolized with t
0.5 < 0.5 h in all examined species. The other 3 examined peptides, i.e., EXE, LIRA, and SEMA, showed higher stability. In particular, LIRA and EXE were shown to be the most stable, with t
0.5 higher than 4 h, followed by SEMA.
3.2. Chemical Degradation of Glutides (GLP-1 RAs) and the Methods Used for Elucidate Their Degradation Pathways
Similarly to low-mass drug (small drug) molecules, regulatory agencies recommend characterizing the chemical stability and related impurities of peptide drugs. These impurities may occur as a result of degradation during manufacture or storage and decrease product efficacy or even exert some toxic effects. It is reported that some impurities derived from the degradation of peptide drugs can even provoke anaphylactic shock [
18]. It is also obvious that impurities from degradation processes in peptide drugs should be controlled using the appropriate strategies [
17].
In the study from the literature [
35], a few analytical methods to examine the degradation of exenatide (EXE) in solutions of different pH were described. Firstly, EXE and its impurities were separated on a C4-Pack column using PDA detection and analyzed by a mass spectrometer with a Q-TOF with a dual ESI system. The total ion chromatogram (TIC) was obtained at 280 nm. Secondly, impurities in EXE were separated using HPLC-SEC using a dedicated Superdex Increase 75 GL column from Cytiva (Marlborough, MA, USA) (10 × 300 mm) and recorded using an UV detector. Samples were separated with phosphate buffered saline as the mobile phase at a flow rate of 1.0 mL/min using isocratic elution. In addition, the intrinsic fluorescence of proteins was used to determine the tertiary structure of EXE. Analysis was performed at the wavelength range of 280–450 nm for emission and at wavelength 270 nm for excitation.
4. Gliflozins (SGLT2 Inhibitors)
Gliflozins are members of a group of relatively new SGLT2 inhibitors that increase glucosuria by inhibiting glucose reabsorption in the kidney. Since the approval of dapagliflozin (DAPA, (2S,3R,4R,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol) as the first SGLT2 inhibitor in 2012, many other drugs have been developed and approved, including canagliflozin (CANA, (2S,3R,4R,5S,6R)-2-[3-[[5-(4-fluorophenyl)thiophen-2-yl]methyl]-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol), empagliflozin (EMPA, (2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol), ertugliflozin (ERTU, (1S,2S,3S,4R,5S)-5-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol), ipragliflozin (IPRA, (2S,3R,4R,5S,6R)-2-[3-(1-benzothiophen-2-ylmethyl)-4-fluorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol), tofogliflozin (TOFO, (3S,3′R,4′S,5′S,6′R)-5-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)spiro [1H-2-benzofuran-3,2′-oxane]-3′,4′,5′-triol), luseogliflozin (LUSE, (2S,3R,4R,5S,6R)-2-[5-[(4-ethoxyphenyl)methyl]-2-methoxy-4-methylphenyl]-6-(hydroxy me thyl)thiane-3,4,5-triol) and bexagliflozin (BEXA, (2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-(2-cyclopropyloxyethoxy)phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol) [
1].
By targeting the kidney, gliflozins have a unique mechanism of action that results in enhanced glucosuria, osmotic diuresis, and natriuresis. Thereby, they improve glucose control with a limited risk of hypoglycemia. In addition, they have shown favorable effects on cardiovascular risk factors such as body weight, blood pressure, lipid profile, arterial stiffness, and endothelial functions [
3,
5]. The predominant pathophysiological mechanisms that may explain the cardiovascular benefits of SGLT2 include plasma volume and diuresis, cardiac fibrosis, myocardial metabolism, and adipokine kinetics. Therefore, the current guidelines propose a new procedure in the management of T2DM with a preferential place for SGLT2, even before metformin, especially in patients with atherosclerotic cardiovascular disease, heart failure, and progressive kidney disease [
1].
4.1. Metabolic Transformations of Gliflozins (SGLT2 Inhibitors) and the Methods Used to Examine Their Metabolic Pathways
In the study of Zhang et al. [
28], the samples from humans and animals receiving bexagliflozin (BEXA) were fractionated by an HPLC system and examined using a radioactivity flow monitor. Radioactivity in bulk samples was determined using a liquid scintillation analyzer. Quench correction was checked using quenched radioactive reference standards. In the next step, unlabeled rat plasma samples were analyzed using the next HPLC system with detection on a triple quadrupole mass spectrometer equipped with an atmospheric pressure chemical ionization (APCI) interface. For the analysis of unlabeled monkey samples, an LC-MS/MS mass spectrometer equipped with a turbo ion spray with an ESI interface was applied.
Based on the results obtained by the Authors, BEXA was shown to be metabolized via glucuronidation and oxidation to form six principal metabolites, i.e., three non-active metabolites through glucuronidation and three through oxidation, i.e., BEXA-M1, BEXA-M2, and BEXA-M3. In vitro studies identified CYP3A4 and UDP-glucuronosyltransferase UGT1A9 as the major enzymes acting on the BEXA moiety. Following oral dosing of humans with [14C]-BEXA, the 3-O-glucuronide contributed 32% of the parent drug, and all other metabolites contributed < 10%. The input of recovered radioactivity was ca. 90%, and of this, ca. 50% was present in feces, predominantly as BEXA, and ca. 40% was present in urine, mainly as 3-O-glucuronide. At the same time, BEXA-M2 was shown as a major metabolite in vivo in all species examined. The pharmacological activity of these metabolites was assessed by measuring the sodium-dependent uptake of the particular substrate, methyl-a-D-[U-14C]-glucopyranoside, that was not metabolized, by cells expressing recombinant human SGLT2 protein. All metabolites had less than 10% of the activity of the parent BEXA [
28].
In the study of Francke et al. [
38], separation of canagliflozin (CANA) and its metabolites was achieved using a Waters XBridge RP-HPLC column (4.6 × 250 mm, 5 µm) at 25 °C. A flow rate of the mobile phase of 1 mL/min was used throughout the analysis. Sample components were eluted with a gradient elution consisting of solvent A (0.025 mM ammonium acetate, pH 9) and solvent B (10/45/45, 0.25 M ammonium acetate of pH 9/MeOH/ACN). The eluates from the HPLC column were steered to both a PDA detector and a radioactivity detector. Finally, the amount of the unchanged CANA was calculated from the radioactivity peaks. The main goal of this study was to examine the formation of CANA O-glucuronides in microsomes from human liver, intestinal, and kidney tissues using [14C]-CANA. Furthermore, the impact of genetic variations in UGTs on the pharmacokinetics of CANA was studied in detail [
38].
Separation of glucuronides of CANA was also performed in the study of Algeelani et al. [
39], using a HPLC method on a Waters μBondaPak C18 column (3.9 × 300 mm) with fluorescence detection. The elution was isocratic, with the mobile phase at pH 3.2 consisting of ACN and 20 mM phosphate buffer (55:45,
v/
v) with a flow rate of 1 mL/min. For detecting respective eluents, the wavelength 280 nm was used for excitation, and the wavelength 325 nm was used for emission.
4.2. Chemical Degradation of Gliflozins (SGLT2 Inhibitors) and the Methods Used for Elucidate Their Degradation Pathways
In the study of Emam and Abdelwaham [
46], CANA was quantified along with its oxidative degradation product by an eco-friendly HPLC method. Separation and quantitation were performed on a Zorbax Eclipse C18 column from Agilent Technologies (4.6 × 250 mm, 5 μm) with the mobile phase consisting of MeOH-H
2O (98:2,
v/
v) at a flow rate of 1 mL/min and UV detection at 225 nm. The column temperature was maintained at 25 °C. The main oxidative degradation product was obtained by refluxing CANA with 3% hydrogen peroxide at high temperatures. The structure of the prepared degradation product showed the presence of a carboxylic group (CANA-D1) that was confirmed using IR analysis [
46].
The degradation processes of CANA were also studied using UPLC and LC/ESI-QTOF-MS/MS analysis [
30]. Separation of the drug and its degradants was monitored at 291 nm. The same UPLC conditions were applied for LC/HRMS analysis. Degradation of CANA was observed under oxidative and acidic stress conditions, whereas it was stable under base and neutral hydrolysis as well as photolytic and thermal stress. The oxidative degradants were identified as CANA-D2 and CANA-D3. Formation of CANA-D2 can be explained by S-oxidation of the thiophene ring and hydroxylation of the fluorobenzene ring of the parent drug, bearing in mind that thiophene sulfur is rather prone to oxidation. The elemental compositions of CANA-D2 and its product ions in MS were confirmed by accurate mass measurements. Based on these data, CANA-D2 was identified as (2S,3R,4S,5S,6R)-2-(3-((5-(4-fluoro-2-hydroxyphenyl)thiophen-1-oxide-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol. As far as CANA-D3 is concerned, the addition of an oxygen atom on the thiophene ring and the formation of 5-(4-fluorophenyl)-2-methylenethiophen-3(2H)-one were suggested. Based on these data, the name for CANA-D3 was proposed as (E)-5-(4-fluorophenyl)-2-(2-methyl-5-((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2Hpyran-2-yl)benzylidene)thiophen-3(2H)-one [
30].
In silico toxicity prediction of DAPA and its degradation products was also performed with ProTox-II software (
https:/tox.charite.de) that classified toxicity with different targets, such as oral, hepatic, carcinogenic, immunotoxicity, mutagenicity, and cytotoxicity. Various Tox21-Nuclear receptor signaling pathways, Tox21-stress response pathways, and toxicity targets were included in this study. Prediction tools identified that there is no binding of DAPA and its degradation products to any of the toxicity targets, and similarly, most of the toxicity end points and pathways are inactive. However, immunotoxicity end point and aryl hydrocarbon receptor signaling pathways are active in the case of DAPA-D2, and mitochondrial membrane potential stress response pathways are active in the cases of DAPA-D2 and DAPA-D1 [
47].
Forced degradation studies were performed under different stress conditions, and then the degraded samples were analyzed by the developed method. In acidic conditions EMPA, showed 28.76% degradation with the formation of 3 degradants, namely, EMPA-D1, EMPA-D2, and EMPA-D3. Based on LC-MS/MS studies, the probable structures of these degradants were proposed. EMPA-D1 was identified as 6-(4-chloro-3-{[4-(oxalan-3yloxy)phenyl]methyl}phenyl)-2-(hydroxymethyl)-3,4-dihydro-2H-pyran-3,4-diol. The structure of 6-(4-chloro-3{[4-(oxalan-3 yloxy)phenyl]methyl}phenyl)-2-(hydroxymethyl)-2H-pyran-3-ol was proposed for EMPA-D2 while EMPA-D3 was specified as 6-(4-chloro-3{[4-(oxalan-3-yloxy)phenyl]methyl}phenyl)-2-methylidene-2H-pyran-3-ol. Under alkaline conditions, EMPA showed degradation of 15.48%, and the formation of the next 2 degradants was reported. From the LC-MS/MS data, the probable structure of one of them, i.e., EMPA-D4 was proposed as 2-(4-hydroxy-3{[4-(oxalan-3-yloxy)phenyl]methyl}phenyl)-6-(hydroxymethyl)-oxane-3,4,5-triol). By analyzing the obtained spectra, the substitution of the chlorine atom by the OH group was proposed. Under oxidative conditions, EMPA was found to generate one more degradant, i.e., EMPA-D5, that could be formed by oxidation of the primary alcoholic group to carboxylic acid to form 6-(4-chloro-3{[4-(oxalan-3-yloxy)phenyl]methyl}phenyl)-3,4,5-trihydroxyoxane-2-carboxylic acid [
49].
5. Conclusions
The main analytical tools for the analysis of glutides and gliflozins are undoubtely LC-MS or LC-HRMS methods. LC-HRMS is especially important for peptide drugs such as glutides. Mass spectrometers are frequently combined with a quadrupole such as a Q-TOF system, an ion trap, or orbitrap mass analyzers that deliver excellent resolution and mass accuracy, which are essential in drug metabolism and drug degradation studies [
15]. In addition, radiometric methods using stable isotopes as well as chromatographic methods with deuterated standards could be used for the determination of the mentioned drugs in biological fluids or excreta. As was described above, [14C] and [13C] technology was widely applied for tracking glutides as well as gliflozins and providing their metabolic profiles.
It was also demonstrated that several SGLT2 inhibitors, including BEXA, CANA, DAPA, EMPA, and ERTU, share a similar metabolic pathway in humans and animals, primarily through O-glucuronidation. Their major non-active metabolites were identified as 1-O, 2-O, and 3-O-glucuronides. These SGLT2 inhibitors that metabolize through glucuronidation can be subsequently eliminated with urine. Such an excretion process can be facilitated by organic anion transporters, which are responsible for the uptake of various organic anionic compounds such as glucuronidation and sulfation drug metabolites [
40].
Furthermore, the more or less important metabolic pathway of SGLT2 inhibitors via oxidative reactions occurred, e.g., for BEXA, DAPA, ERTU, LUSE, and TOFO. Such metabolic pathways, e.g., ω-hydroxylation at the ethoxy group followed by oxidation to form the corresponding carboxylic acid, were shown for LUSE, leading to the formation of LUSE-M4. The metabolic pathway via oxidation of the substituents at the 4-phenyl position of BEXA, DAPA, ERTU, LUSE, and TOFO seems to also be the characteristic pattern of their metabolism. The hydroxyl metabolite TOFO-M1 undergoes further oxidation to the respective ketone metabolite TOFO-M2. The next hydroxyl metabolite, TOFO-M3, is further oxidized to the corresponding phenyl acetic acid metabolite, TOFO-M4.
Generally, both drug metabolism and drug degradation (during drug manufacturing and/or storage) can undergo similar chemical transformations. Consequently, many impurities that are generated during degradation could also be respective metabolites. Sometimes, individual metabolites as well as particular degradation products are formed or detected in small amounts, potentially with no toxicity risk. However, the goal of the present paper was to find as many such overlapping products of metabolism and chemical degradation of gliflozins as possible Thus, the metabolites and degradants formed in smaller amounts were also included. Further studies on possible degradation products of gliflozins based on their metabolic pathways should give a definitive assessment of their safety for patients. As far as such detected similarities between metabolites and degradants are concerned, the process of hydroxylation of the benzyl-phenyl or benzyl-thiophen moieties that was characteristic for metabolites of BEXA-M3, CANA-M2, DAPA-M1 and ERTU-M1 was observed during degradation of EMPA to form EMPA-D6.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11082127