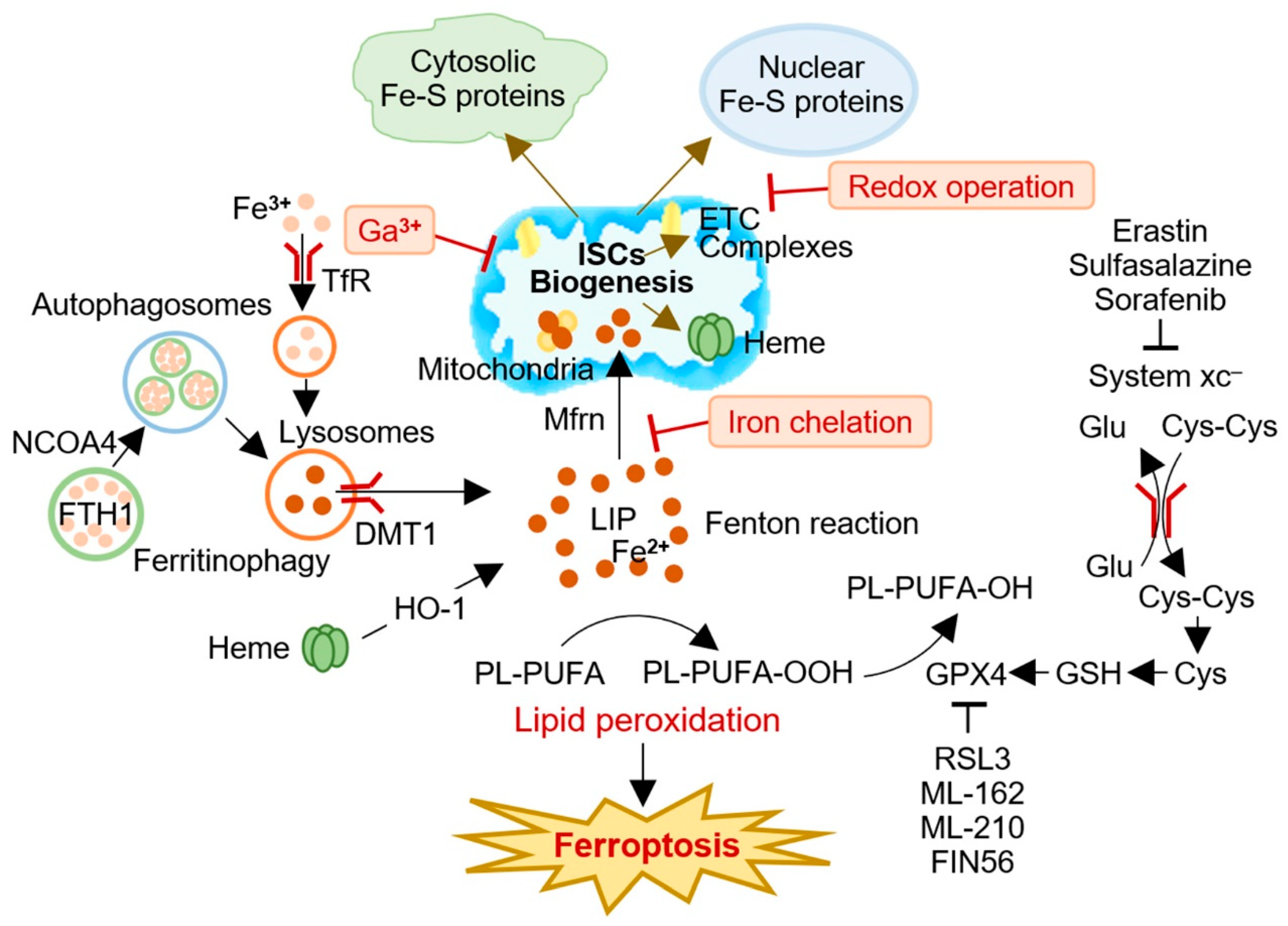
Iron dysregulation is a hallmark of cancer, characterized by an overexpression of genes involved in iron metabolism and iron-sulfur cluster (ISC) biogenesis. Dysregulated iron homeostasis increases intracellular labile iron, which may lead to the formation of excess cytotoxic radicals and make it vulnerable to various types of regulated cell death, including ferroptosis. The inhibition of ISC synthesis triggers the iron starvation response, increasing lipid peroxidation and ferroptosis in cancer cells treated with oxidative stress-inducing agents. Various methods, such as redox operations, iron chelation, and iron replacement with redox-inert metals, can destabilize or limit ISC formation and function, providing potential therapeutic strategies for cancer treatment. Targeting ISCs to induce ferroptosis represents a promising approach in cancer therapy.
- iron
- ferroptosis
- iron-sulfur cluster
- cancer
- therapy
1. Iron Metabolism and Ferroptosis in Cancer
2. ISC Modulation and Ferroptosis in Cancer
3. Implications of Targeting ISCs for Ferroptosis-Based Cancer Therapy
This entry is adapted from the peer-reviewed paper 10.3390/cancers15102694
References
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125.
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455.
- Basak, T.; Kanwar, R.K. Iron imbalance in cancer: Intersection of deficiency and overload. Cancer Med. 2022, 11, 3837–3853.
- Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An Essential Element of Cancer Metabolism. Cells 2020, 9, 2591.
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021.
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226.
- Gao, G.; Zhang, N.; Wang, Y.Q.; Wu, Q.; Yu, P.; Shi, Z.H.; Duan, X.L.; Zhao, B.L.; Wu, W.S.; Chang, Y.Z. Mitochondrial Ferritin Protects Hydrogen Peroxide-Induced Neuronal Cell Damage. Aging Dis. 2017, 8, 458–470.
- Chiang, S.; Kovacevic, Z.; Sahni, S.; Lane, D.J.; Merlot, A.M.; Kalinowski, D.S.; Huang, M.L.; Richardson, D.R. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clin. Sci. 2016, 130, 853–870.
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792.
- Venkataramani, V. Iron Homeostasis and Metabolism: Two Sides of a Coin. Adv. Exp. Med. Biol. 2021, 1301, 25–40.
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897.
- Kurz, T.; Eaton, J.W.; Brunk, U.T. The role of lysosomes in iron metabolism and recycling. Int. J. Biochem. Cell Biol. 2011, 43, 1686–1697.
- Tabuchi, M.; Yoshimori, T.; Yamaguchi, K.; Yoshida, T.; Kishi, F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 2000, 275, 22220–22228.
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell Biol. 2011, 31, 2040–2052.
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic. Biol. Med. 2011, 50, 1647–1658.
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006, 273, 3106–3117.
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109.
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102.
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238.
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv. Exp. Med. Biol. 2021, 1301, 41–57.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203.
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285.
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308.
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726.
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The role of iron in cancer progression. Front. Oncol. 2021, 11, 778492.
- Alkhateeb, A.A.; Connor, J.R. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim. Biophys. Acta 2013, 1836, 245–254.
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588.
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39.
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Goswami, P.C.; Griguer, C.E. Cytochrome c oxidase mediates labile iron level and radioresistance in glioblastoma. Free Radic. Biol. Med. 2022, 185, 25–35.
- Gupta, P.; Singh, P.; Pandey, H.S.; Seth, P.; Mukhopadhyay, C.K. Phosphoinositide-3-kinase inhibition elevates ferritin level resulting depletion of labile iron pool and blocking of glioma cell proliferation. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 547–564.
- Petronek, M.S.; Spitz, D.R.; Allen, B.G. Iron-Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants 2021, 10, 1458.
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763.
- Banci, L.; Camponeschi, F.; Ciofi-Baffoni, S.; Piccioli, M. The NMR contribution to protein-protein networking in Fe-S protein maturation. J. Biol. Inorg. Chem. 2018, 23, 665–685.
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499.
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.; Linehan, W.M.; Rouault, T.A. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311.
- Wang, S.; Liu, Z.; Geng, J.; Li, L.; Feng, X. An overview of ferroptosis in non-alcoholic fatty liver disease. Biomed. Pharmacother. 2022, 153, 113374.
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.T.; Lipper, C.H.; Morcos, F.; et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 2015, 1853, 1294–1315.
- Sohn, Y.S.; Tamir, S.; Song, L.; Michaeli, D.; Matouk, I.; Conlan, A.R.; Harir, Y.; Holt, S.H.; Shulaev, V.; Paddock, M.L.; et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 14676–14681.
- Karmi, O.; Sohn, Y.S.; Zandalinas, S.I.; Rowland, L.; King, S.D.; Nechushtai, R.; Mittler, R. Disrupting CISD2 function in cancer cells primarily impacts mitochondrial labile iron levels and triggers TXNIP expression. Free Radic. Biol. Med. 2021, 176, 92–104.
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844.
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox. Signal. 2019, 30, 1083–1095.
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190.
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.S.; Jennings, P.A.; et al. Interactions between mitoNEET and NAF-1 in cells. PLoS ONE 2017, 12, e0175796.
- Li, Y.; Xu, B.; Ren, X.; Wang, L.; Xu, Y.; Zhao, Y.; Yang, C.; Yuan, C.; Li, H.; Tong, X.; et al. Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell. Mol. Biol. Lett. 2022, 27, 81.
- Li, Y.; Wang, X.; Huang, Z.; Zhou, Y.; Xia, J.; Hu, W.; Wang, X.; Du, J.; Tong, X.; Wang, Y. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2022, 27, 839.
- Jiang, H.; Fang, Y.; Wang, Y.; Li, T.; Lin, H.; Lin, J.; Pan, T.; Liu, Q.; Lv, J.; Chen, D.; et al. FGF4 improves hepatocytes ferroptosis in autoimmune hepatitis mice via activation of CISD3. Int. Immunopharmacol. 2023, 116, 109762.
- Tsaousis, A.D. On the Origin of Iron/Sulfur Cluster Biosynthesis in Eukaryotes. Front. Microbiol. 2019, 10, 2478.
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643.
- Chafe, S.C.; Vizeacoumar, F.S.; Venkateswaran, G.; Nemirovsky, O.; Awrey, S.; Brown, W.S.; McDonald, P.C.; Carta, F.; Metcalfe, A.; Karasinska, J.M.; et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci. Adv. 2021, 7, eabj0364.
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal. Transduct. Target Ther. 2022, 7, 54.
- Fujihara, K.M.; Zhang, B.Z.; Jackson, T.D.; Ogunkola, M.O.; Nijagal, B.; Milne, J.V.; Sallman, D.A.; Ang, C.S.; Nikolic, I.; Kearney, C.J.; et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci. Adv. 2022, 8, eabm9427.
- Prischi, F.; Konarev, P.V.; Iannuzzi, C.; Pastore, C.; Adinolfi, S.; Martin, S.R.; Svergun, D.I.; Pastore, A. Structural bases for the interaction of frataxin with the central components of iron-sulphur cluster assembly. Nat. Commun. 2010, 1, 95.
- Pandolfo, M.; Pastore, A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 2009, 256 (Suppl. S1), 9–17.
- Guccini, I.; Serio, D.; Condò, I.; Rufini, A.; Tomassini, B.; Mangiola, A.; Maira, G.; Anile, C.; Fina, D.; Pallone, F.; et al. Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis. 2011, 2, e123.
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox. Biol. 2020, 32, 101483.
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 2018, 10, 49–72.
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox. Biol. 2021, 43, 101975.
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761.
- Liu, G.; Wang, Y.; Anderson, G.J.; Camaschella, C.; Chang, Y.; Nie, G. Functional Analysis of GLRX5 Mutants Reveals Distinct Functionalities of GLRX5 Protein. J. Cell Biochem. 2016, 117, 207–217.
- Lee, J.; You, J.H.; Shin, D.; Roh, J.L. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786.
- Green, Y.S.; Ferreira Dos Santos, M.C.; Fuja, D.G.; Reichert, E.C.; Campos, A.R.; Cowman, S.J.; Acuña Pilarte, K.; Kohan, J.; Tripp, S.R.; Leibold, E.A.; et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene 2022, 41, 4709–4723.
- Wu, H.; Wei, H.; Zhang, D.; Sehgal, S.A.; Zhang, D.; Wang, X.; Qin, Y.; Liu, L.; Chen, Q. Defective mitochondrial ISCs biogenesis switches on IRP1 to fine tune selective mitophagy. Redox. Biol. 2020, 36, 101661.
- Terzi, E.M.; Sviderskiy, V.O.; Alvarez, S.W.; Whiten, G.C.; Possemato, R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021, 7, eabg4302.
- Ravichandran, M.; Hu, J.; Cai, C.; Ward, N.P.; Venida, A.; Foakes, C.; Kuljanin, M.; Yang, A.; Hennessey, C.J.; Yang, Y.; et al. Coordinated Transcriptional and Catabolic Programs Support Iron-Dependent Adaptation to RAS-MAPK Pathway Inhibition in Pancreatic Cancer. Cancer Discov. 2022, 12, 2198–2219.
- Santana-Codina, N.; Del Rey, M.Q.; Kapner, K.S.; Zhang, H.; Gikandi, A.; Malcolm, C.; Poupault, C.; Kuljanin, M.; John, K.M.; Biancur, D.E.; et al. NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron-Sulfur Cluster Proteins. Cancer Discov. 2022, 12, 2180–2197.
- Zhang, M.; Liu, Z.; Le, Y.; Gu, Z.; Zhao, H. Iron-Sulfur Clusters: A Key Factor of Regulated Cell Death in Cancer. Oxid. Med. Cell Longev. 2022, 2022, 7449941.
- Weon, J.L.; Yang, S.W.; Potts, P.R. Cytosolic Iron-Sulfur Assembly Is Evolutionarily Tuned by a Cancer-Amplified Ubiquitin Ligase. Mol. Cell. 2018, 69, 113–125.e6.
- Lelièvre, P.; Sancey, L.; Coll, J.L.; Deniaud, A.; Busser, B. Iron Dysregulation in Human Cancer: Altered Metabolism, Biomarkers for Diagnosis, Prognosis, Monitoring and Rationale for Therapy. Cancers 2020, 12, 3524.
- Propper, R.D.; Cooper, B.; Rufo, R.R.; Nienhuis, A.W.; Anderson, W.F.; Bunn, H.F.; Rosenthal, A.; Nathan, D.G. Continuous subcutaenous administration of deferoxamine in patients with iron overload. N. Engl. J. Med. 1977, 297, 418–423.
- Sandoval-Acuña, C.; Torrealba, N.; Tomkova, V.; Jadhav, S.B.; Blazkova, K.; Merta, L.; Lettlova, S.; Adamcová, M.K.; Rosel, D.; Brábek, J.; et al. Targeting Mitochondrial Iron Metabolism Suppresses Tumor Growth and Metastasis by Inducing Mitochondrial Dysfunction and Mitophagy. Cancer Res. 2021, 81, 2289–2303.
- Estrov, Z.; Tawa, A.; Wang, X.H.; Dubé, I.D.; Sulh, H.; Cohen, A.; Gelfand, E.W.; Freedman, M.H. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987, 69, 757–761.
- Donfrancesco, A.; Deb, G.; Dominici, C.; Pileggi, D.; Castello, M.A.; Helson, L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990, 50, 4929–4930.
- Dreicer, R.; Kemp, J.D.; Stegink, L.D.; Cardillo, T.; Davis, C.S.; Forest, P.K.; See, W.A. A phase II trial of deferoxamine in patients with hormone-refractory metastatic prostate cancer. Cancer Investig. 1997, 15, 311–317.
- Bedford, M.R.; Ford, S.J.; Horniblow, R.D.; Iqbal, T.H.; Tselepis, C. Iron chelation in the treatment of cancer: A new role for deferasirox? J. Clin. Pharmacol. 2013, 53, 885–891.
- Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529.
- Tong, W.H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron-sulfur cluster biogenesis. Biometals 2007, 20, 549–564.
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662.
- Gęgotek, A.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Activity of Ascorbic Acid. Antioxidants 2022, 11, 1993.
- Liu, Y.; Liu, C.; Li, J. Comparison of Vitamin C and Its Derivative Antioxidant Activity: Evaluated by Using Density Functional Theory. ACS Omega 2020, 5, 25467–25475.
- Szarka, A.; Kapuy, O.; Lőrincz, T.; Bánhegyi, G. Vitamin C and Cell Death. Antioxid. Redox. Signal. 2021, 34, 831–844.
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e488.
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218.
- Wang, X.; Xu, S.; Zhang, L.; Cheng, X.; Yu, H.; Bao, J.; Lu, R. Vitamin C induces ferroptosis in anaplastic thyroid cancer cells by ferritinophagy activation. Biochem Biophys. Res. Commun. 2021, 551, 46–53.
- Tang, H.M.; Tang, H.L. Cell recovery by reversal of ferroptosis. Biol. Open 2019, 8, bio043182.
- Chitambar, C.R. The therapeutic potential of iron-targeting gallium compounds in human disease: From basic research to clinical application. Pharmacol. Res. 2017, 115, 56–64.
- Chitambar, C.R. Gallium and its competing roles with iron in biological systems. Biochim. Biophys. Acta 2016, 1863, 2044–2053.
- Chitambar, C.R. Gallium Complexes as Anticancer Drugs. Met. Ions Life Sci. 2018, 18, 469–506.
- Straus, D.J. Gallium nitrate in the treatment of lymphoma. Semin. Oncol. 2003, 30, 25–33.
- Bernstein, L.R.; Tanner, T.; Godfrey, C.; Noll, B. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Met. Based Drugs 2000, 7, 33–47.
- Chitambar, C.R.; Al-Gizawiy, M.M.; Alhajala, H.S.; Pechman, K.R.; Wereley, J.P.; Wujek, R.; Clark, P.A.; Kuo, J.S.; Antholine, W.E.; Schmainda, K.M. Gallium Maltolate Disrupts Tumor Iron Metabolism and Retards the Growth of Glioblastoma by Inhibiting Mitochondrial Function and Ribonucleotide Reductase. Mol. Cancer Ther. 2018, 17, 1240–1250.
- Li, H.; Stümpfig, M.; Zhang, C.; An, X.; Stubbe, J.; Lill, R.; Huang, M. The diferric-tyrosyl radical cluster of ribonucleotide reductase and cytosolic iron-sulfur clusters have distinct and similar biogenesis requirements. J. Biol. Chem. 2017, 292, 11445–11451.
- Nagai, S.; Yoshida, M.; Takigawa, Y.; Torii, S.; Koshiishi, I. Botanical sulfane sulfur donors inhibit ferroptotic cell death caused by the depletion of cysteine. Food Chem. 2021, 343, 128511.
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369.