Adult stem cells lie at the crossroads of tissue repair, inflammation, and malignancy. Intestinal microbiota and microbe–host interactions are pivotal to maintaining gut homeostasis and response to injury, and participate in colorectal carcinogenesis. Yet, limited knowledge is available on whether and how bacteria directly crosstalk with intestinal stem cells (ISC), particularly cancerous stem-like cells (CR-CSC), as engines for colorectal cancer initiation, maintenance, and metastatic dissemination. Among several bacterial species alleged to initiate or promote colorectal cancer (CRC), the pathobiont Fusobacterium Nucleatum has recently drawn significant attention for its epidemiologic association and mechanistic linkage with the disease.
- colorectal cancer
- Fusobacterium nucleatum
- Helicobacter Pylori
- intestinal stem cells
1. Bacterial Carcinogenesis in Colorectal Cancer
2. Bacteria and CRC: Intestinal Repair Gone Awry?
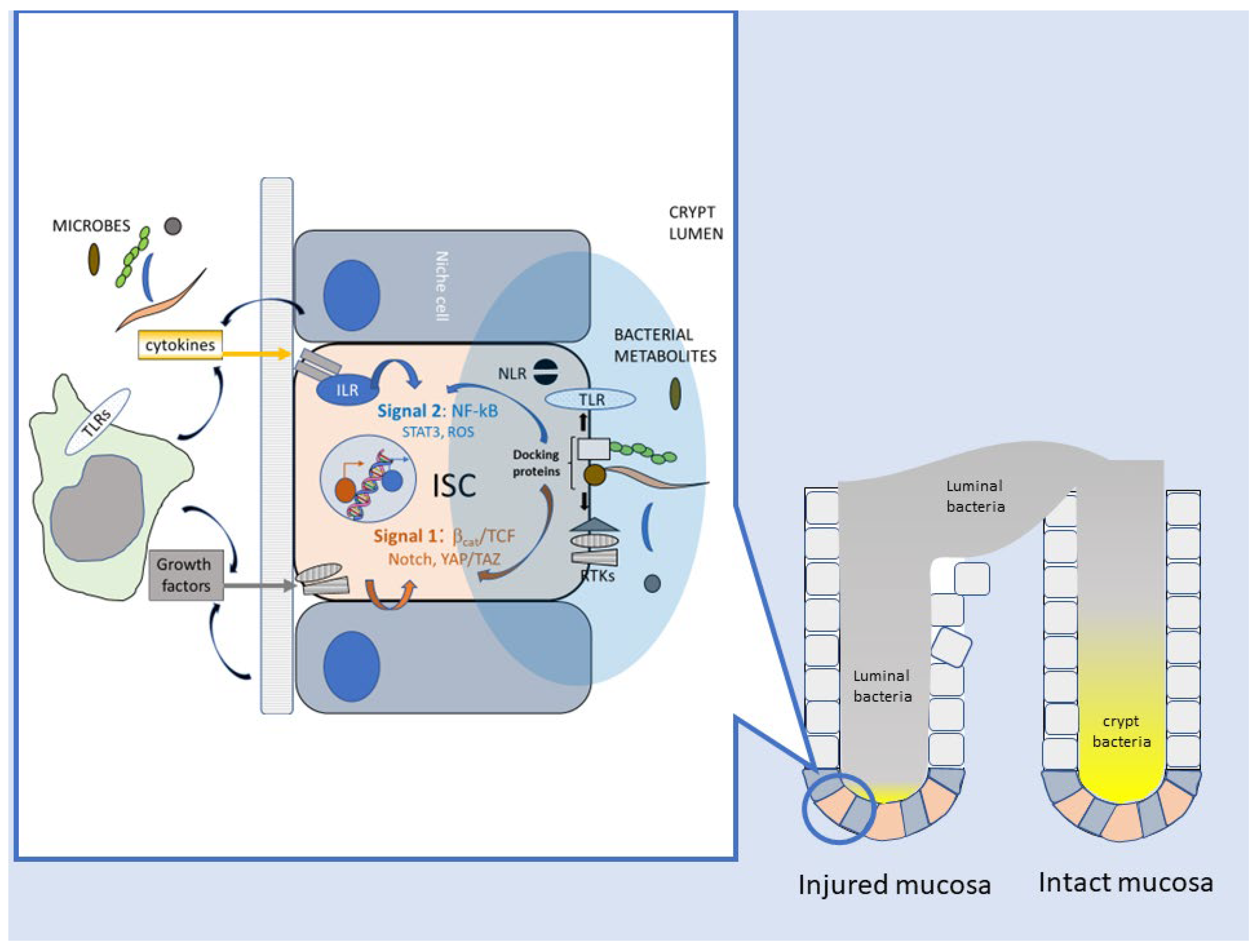
2.1. Enhancement of Stem-like Features in Epithelial Cells
2.2. Reparative Inflammatory Responses
2.3. Downregulation of Adaptive Immunity
This entry is adapted from the peer-reviewed paper 10.3390/cancers15092583
References
- Greig, J.M.; Ellis, C.J. Biological Agents. In Occupational Hygiene, 3rd ed.; Wiley Online Library: Hoboken, NJ, USA, 2008; Volume 100, pp. 344–359.
- Hamid, H.K.S. Schistosoma Japonicum–Associated Colorectal Cancer: A Review. Am. J. Trop. Med. Hyg. 2019, 100, 501.
- Aries, V.; Crowther, J.S.; Drasar, B.S.; Hill, M.J.; Williams, R.E. Bacteria and the Aetiology of Cancer of the Large Bowel. Gut 1969, 10, 334–335.
- Elsland, D.; Neefjes, J. Bacterial Infections and Cancer. EMBO Rep. 2018, 19, e46632.
- Sears, C.L.; Garrett, W.S. Microbes, Microbiota, and Colon Cancer. Cell Host Microbe 2014, 15, 317–328.
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J. Infect. Dis. 2011, 203, 306–311.
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A Bacterial Driver-Passenger Model for Colorectal Cancer: Beyond the Usual Suspects. Nat. Rev. Microbiol. 2012, 10, 575–582.
- Garrett, W.S. Cancer and the Microbiota. Science 2015, 348, 80–86.
- Hatakeyama, M.; Higashi, H. Helicobacter Pylori CagA: A New Paradigm for Bacterial Carcinogenesis. Cancer Sci. 2005, 96, 835–843.
- Song, X.; Xin, N.; Wang, W.; Zhao, C. Wnt/β-Catenin, an Oncogenic Pathway Targeted by H. Pylori in Gastric Carcinogenesis. Oncotarget 2015, 6, 35579.
- Pöltl, L.; Kitsera, M.; Raffl, S.; Schild, S.; Cosic, A.; Kienesberger, S.; Unterhauser, K.; Raber, G.; Lembacher-Fadum, C.; Breinbauer, R.; et al. Microbiota-Derived Genotoxin Tilimycin Generates Colonic Stem Cell Mutations. Cell Rep. 2023, 42.
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with Familial Adenomatous Polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science 2018, 359, 592–597.
- Hatakeyama, M. Helicobacter Pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306–316.
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum Infection Is Prevalent in Human Colorectal Carcinoma. Genome Res. 2012, 22, 299–306.
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic Analysis Identifies Association of Fusobacterium with Colorectal Carcinoma. Genome Res. 2012, 22, 292–298.
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach. Nature 2020, 579, 567–574.
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38.
- Houghton, J.M.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric Cancer Originating from Bone Marrow-Derived Cells. Science 2004, 306, 1568–1571.
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 Immunity in Tissue Repair and Fibrosis. Nat. Rev. Immunol. 2017, 18, 62–76.
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462.
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Liu, H.; Du, J.; Chao, S.; Li, S.; Cai, H.; Zhang, H.; Chen, G.; Liu, P.; Bu, P. Fusobacterium nucleatum Promotes Colorectal Cancer Cell to Acquire Stem Cell-Like Features by Manipulating Lipid Droplet-Mediated Numb Degradation. Adv. Sci. 2022, 9, 2105222.
- Lee, D.G.; Kim, H.S.; Lee, Y.S.; Kim, S.; Cha, S.Y.; Ota, I.; Kim, N.H.; Cha, Y.H.; Yang, D.H.; Lee, Y.; et al. Helicobacter Pylori CagA Promotes Snail-Mediated Epithelial–mesenchymal Transition by Reducing GSK-3 Activity. Nat. Commun. 2014, 5, 4423.
- Lu, R.; Wu, S.; Zhang, Y.G.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric Bacterial Protein AvrA Promotes Colonic Tumorigenesis and Activates Colonic Beta-Catenin Signaling Pathway. Oncogenesis 2014, 3, e105.
- Cavallucci, V.; Palucci, I.; Fidaleo, M.; Mercuri, A.; Masi, L.; Emoli, V.; Bianchetti, G.; Fiori, M.E.; Bachrach, G.; Scaldaferri, F.; et al. Proinflammatory and Cancer-Promoting Pathobiont Fusobacterium nucleatum Directly Targets Colorectal Cancer Stem Cells. Biomolecules 2022, 12, 1256.
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter Pylori Activates and Expands Lgr5+ Stem Cells through Direct Colonization of the Gastric Glands. Gastroenterology 2015, 148, 1392–1404.e21.
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206.
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umanã, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum Host-Cell Binding and Invasion Induces IL-8 and CXCL1 Secretion That Drives Colorectal Cancer Cell Migration. Sci. Signal. 2020, 13, eaba9157.
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 Promotes Intestinal-Stem-Cell-Mediated Epithelial Regeneration. Nature 2015, 528, 560–564.
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; De Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A Gp130–Src–YAP Module Links Inflammation to Epithelial Regeneration. Nature 2015, 519, 57–62.
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402.
- Volonté, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-Initiating Cells from Colorectal Cancer Patients Escape from T Cell–Mediated Immunosurveillance In Vitro through Membrane-Bound IL-4. J. Immunol. 2014, 192, 523–532.
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS Production and NF-ΚB Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell 2013, 12, 761–773.
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296.
- Wizenty, J.; Müllerke, S.; Kolesnichenko, M.; Heuberger, J.; Lin, M.; Fischer, A.; Mollenkopf, H.; Berger, H.; Tacke, F.; Sigal, M. Gastric Stem Cells Promote Inflammation and Gland Remodeling in Response to Helicobacter Pylori via Rspo3-Lgr4 Axis. EMBO J. 2022, 41, e109996.
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619.
- Schmitz, M.L.; Krappmann, D. Controlling NF-ΚB Activation in T Cells by Costimulatory Receptors. Cell Death Differ. 2006, 13, 834–842.
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355.
- Galaski, J.; Shhadeh, A.; Umaña, A.; Yoo, C.C.; Arpinati, L.; Isaacson, B.; Berhani, O.; Singer, B.B.; Slade, D.J.; Bachrach, G.; et al. Fusobacterium nucleatum CbpF Mediates Inhibition of T Cell Function Through CEACAM1 Activation. Front. Cell. Infect. Microbiol. 2021, 11, 692544.
- Fournier, B.M.; Parkos, C.A. The Role of Neutrophils during Intestinal Inflammation. Mucosal Immunol. 2012, 5, 354–366.
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The Regulatory Roles of Neutrophils in Adaptive Immunity. Cell Commun. Signal. 2019, 17, 147.
- Teijeira, A.; Garasa, S.; Ochoa, M.C.; Villalba, M.; Olivera, I.; Cirella, A.; Eguren-Santamaria, I.; Berraondo, P.; Schalper, K.A.; de Andrea, C.E.; et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin. Cancer Res. 2021, 27, 2383–2393.