Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Bradykinin (BK) metabolism and its receptors play a central role in drug-induced angioedema (AE) without urticaria through increased vascular permeability. Many cardiovascular and diabetic drugs may cause BK-mediated AE. Angiotensin-converting enzyme inhibitors (ACEIs) and neprilysin inhibitors impair BK catabolism.
- bradykinin metabolism
- drug-induced angioedema
1. Introduction
Angioedema (AE) is a transient subcutaneous or submucosal swelling that is non-pitting when pressure is applied. It differs from edema and is caused by an accumulation of fluid in the interstitium and characterized by persistent pitting with pressure. AE is a presenting manifestation that results from an underlying pathophysiologic process involving the localized or systemic release of vasoactive mediators, most frequently histamine or bradykinin (BK) [1][2]. Differentiating between histaminergic AE and BK-mediated AE is of utmost importance, not only due to the different treatment approaches but also because the latter can be more often life-threatening. Acute episodes of AE without accompanying urticaria characterize BK-mediated AE, and these do not respond to conventional treatment with systemic H1 antihistamines, corticosteroids, or adrenaline. BK-mediated AE may be hereditary, acquired, or drug-induced [1][3][4][5].
2. Overview on Epidemiology of BK-Mediated AE
The overall incidence of AE induced by angiotensin-converting enzyme inhibitors (ACEI-AE), the most important cause of BK-mediated AE, is less than 1%. Still, this therapeutic class is the leading cause of drug-induced AE, accounting for up to 40% of emergency room visits for AE, with relatively constant and persistent annual risk [6]. While AE that occurs soon after starting the ACEI treatment is common, many cases occur more than six weeks after initiating therapy. In addition, cases of late-onset angioedema are reported up to 23 years after the initiation of ACEI administration. Thus, AE can occur at any time during the treatment with an ACEI [4][7]. Recently it was estimated that the average duration of ACEI use among patients presenting with AE exceeds two years [8]. Such a time course represents a problem for the healthcare provider and the patient; for the clinician, the diagnosis may be difficult due to not recognizing the association, whereas for the misdiagnosed patient, the delay prolongs a potentially dangerous situation. ACEI-AE is more common (0.1–0.7% of treated patients) than other rare BK-mediated AE, such as hereditary angioedema (HAE), due to complement C1 esterase inhibitor (C1-INH) deficiency (1:10,000–1:50,000 prevalence), which is more common than acquired angioedema with C1 inhibitor deficiency (1:100,000 to 1:500,000 prevalence) [2][9][10].
3. Diagnostic Approach in Drug-Induced BK-Mediated AE
Currently, the diagnosis of drug-induced AE mediated by BK is clinical. There are no available laboratory tests to diagnose this non-allergic adverse reaction routinely. However, resolution following the discontinuation of the drug confirms the diagnosis.
ACEI-induced AE most often involves the head, neck, face, lips, tongue, and larynx. Rarely, it affects visceral organs. Life-threatening edema of the upper airway presents in about 25–40% of cases of ACEI-AE. ACEIs are the most common cause of BK-mediated AE, which can be life-threatening.
BK-mediated AE episodes induced by antihypertensive drugs acting on the renin–angiotensin–aldosterone system (RAAS), such as ACEIs, blockers of angiotensin II type 1 receptors (ARBs), and direct renin inhibitors (DRIs), primarily affect the head and neck region and may occur even after several years of uneventful treatment. Diagnosis is clinical but not easy for clinicians unfamiliar with such an adverse reaction, as the appearance resembles allergic AE. No specific diagnostic biomarkers are available, and no officially approved treatment is currently accessible [11].
Awareness of the epidemiology and risk factors for ACEI-AE is critical for appropriately managing any patient treated with ACEI, because it is associated with high morbidity (intubation, intensive care unit admissions, and hospitalizations) and fatal reactions have been documented [6]. BK-mediated AE can occur due to vasodilation and increased vascular permeability, mainly due to the inflammatory mediator BK. This form of AE can result from either overproduction of BK or, more frequently, from inhibition of BK degradation. Pruritus and urticaria are absent, mast cells being not involved, and it most commonly affects the lips, tongue, face, and upper airways. The intestine may also be involved, presenting as acute abdominal pain with diarrhea or other gastrointestinal symptoms, but this presentation may be less usually recognized. The reason why certain regions of the body are more often affected has yet to be comprehended [1][8].
4. Metabolism of BK and Its Receptors
The vasoactive nonapeptides BK and kallidin (Lys-BK) exert their bioactivities by binding to constitutively expressed kinin B2 receptors before being metabolized by multiple peptidases. In various cells and tissues, neutral endopeptidase (NEP, neprilysin) plays a significant role in the degradation of BK. In human plasma, BK is metabolized mostly by three metallopeptidases. Angiotensin I-converting enzyme (ACE) constitutes the main degradation pathway. Carboxypeptidase N (CPN) transforms BK and kallidin into active metabolites, des-arginine9-BK and Lys-des-Arg9-BK, respectively. This is a minor metabolic plasmatic pathway unless ACE is inhibited. Also, aminopeptidase P (X-pro aminopeptidase, proline aminopeptidase, APP) plays an important role in the metabolism of kinins in plasma, mostly for des-Arg9-BK. Although the carboxy-truncated metabolites of BK are largely inactive under normal conditions, but may be agonists of inflammation-regulated kinin B1 receptors [1][9][12]. By assessing the gene expression of metallopeptidases and kinin receptors in the oropharyngeal pig tissues, it was revealed that B2 receptor regulation might be modulated in a tissue-specific manner through ACEI treatment with mRNA upregulation in the tongue and laryngeal tissues [12].
In an overview of the renin–angiotensin–aldosterone system (RAAS), renin is secreted in the kidney by the juxtaglomerular cells in response to decreased renal perfusion pressures; the precursor angiotensinogen produced in the liver is converted by renin to produce the weak vasoconstrictor angiotensin I, which is further metabolized in the lungs by the ACE to create the potent vasoconstrictor angiotensin II (ATII), which acts on vascular endothelial receptors. The two types of ATII receptors, AT1 and AT2 receptors, which are present in the heart and vasculature smooth muscle, mediate its vasoconstrictive action. In addition, it also acts on the zona glomerulosa of the adrenal cortex to stimulate the release of aldosterone. This steroid hormone acts on the kidneys to promote sodium and water retention. Therefore, ACEI effects result from the inhibition of ACE, ultimately impacting the RAAS cascade and causing a decrease in systemic blood pressure [7][13].
Kinins are a group of active peptides released into body fluids and tissues following the enzymatic action of kallikreins on kininogens, which occurs through a complex proteolytic cascade of events called the kallikrein–kinin cascade. This contact activation pathway is initiated when factor XII (Hageman factor) binds to damaged tissue, becoming activated through conversion to factor XIIa. This converts prekallikrein to plasma kallikrein, and these two proteins autoactivate each other through a positive feedback loop. Plasma kallikrein cleaves high-molecular-weight kininogen (HMWK), producing BK. The binding of BK to its B2 receptors induces vasodilation and increased endothelial permeability, generating the characteristic manifestations of an acute episode of AE [2].
BK is generated through the cleavage of HMWK by activated kallikrein, which occurs through the contact system via factor XII, which can be activated by plasmin through the fibrinolysis pathway. In addition, the C1 esterase inhibitor (C1-INH), which acts as a brake on the complement system, blocks BK overproduction via the contact system [14][15].
ACE (also known as kininase II) plays a significant role in the renin–angiotensin–aldosterone system through two proteolytic mechanisms: conversion of angiotensin I to angiotensin II, and degradation of BK. These two actions make ACE inhibition a principal target in the treatment of hypertension, myocardial infarction (MI), heart failure, and type I diabetic nephropathy [2]. ACE is a crucial peptidase involved in the degradation of the vasoactive nonapeptide BK with a short half-life (about 17 s) to inactive metabolites. BK stimulates constitutively expressed B2 receptors, thus inducing vasodilation and vascular permeability and stimulating the release of SP from sensory fibers with further increased vascular permeability [16][17][18].
The amino acid sequence of BK is presented in Figure 1, along with the sites of cleavage by different enzymes significant in the pathogenesis of drug-induced BK-mediated AE. By focusing on these enzymes, clinicians can better comprehend the complexities of BK-related drug-induced AE. Three specific metalloproteinases are involved in the primary degradation pathways of BK: ACE, APP, and NEP. Secondary enzymes include dipeptidyl peptidase-4 (DPP-IV) and CPN (kininase I).
ACEIs rapidly and strongly reduce ACE activity, but only weakly affect APP and NEP [13][19]. When ACE is inhibited by ACEIs, other BK metabolic enzymes, APP and NEP, along with DPP-4 and CPN, play a more significant role in degrading BK, des-Arg9-BK, and SP. These three mediators play critical roles in the pathophysiology of ACEI-AE, with BK having a central role in this BK-mediated AE. A secondary role is played by des-Arg9-BK, an active metabolite of BK formed primarily due to the CPN. The activities of the des-Arg9-BK, a selective agonist of the inducible B1 type receptors for kinins, are also short-lived because of its breakdown by APP, DPP-4, and ACE2. SP, inactivated primarily by the DPP-4 but also through NEP and ACE, also plays a secondary role. BK, des-Arg9-BK, and SP are challenging to measure due to their extremely short half-lives and the variability between serum and tissue concentrations in different tissues. Inhibition of ACE by ACEI impacts the degradation pathways of these compounds, and their elevated levels are associated with AE; however, multiple and redundant enzyme pathways may be involved [13][20][21][22].
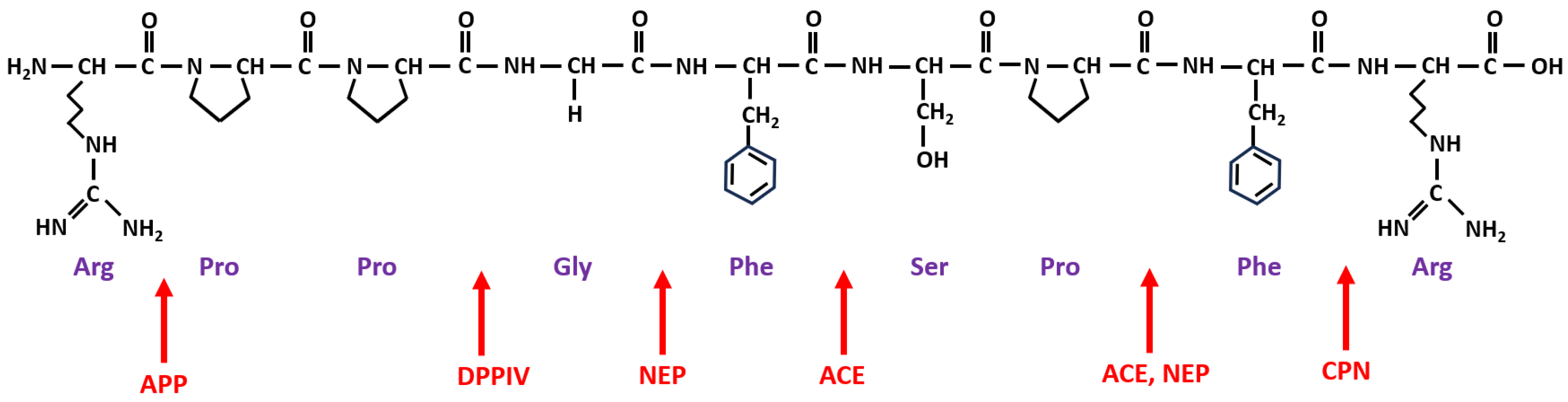
Figure 1. Structure of BK and sites of cleavage by significant enzymes related to drug-induced AE. Note: The amino acid sequence of BK is Arginine (Arg)–Proline (Pro)–Proline (Pro)–Glycine (Gly)–Phenylalanine (Phe)–Serine (Ser)–Proline (Pro)–Phenylalanine (Phe)–Arginine (Arg). A simplified presentation of the sites of cleavage of BK by different important enzymes, marked with red arrows for ACE (angiotensin-converting enzyme), APP (aminopeptidase P), NEP (neutral endopeptidase), CPN (carboxypeptidase N), and DPPIV (dipeptidyl peptidase IV).
Defects or deficiencies in enzymes controlling BK catabolism, such as reduced APP activity, decreased DPP-4 antigen and activity levels, and slower degradation of des-Arg9-BK, predispose patients to develop AE when taking an ACEI [20][21][22]. Unlike patients with C1-INH deficiency, those with ACEI-AE show no evidence of the cleavage products of HMWK in their plasma, despite high plasma concentrations of BK. Because the cleavage of HMWK generates BK, the pathogenic mechanism of ACEI-AE resides in the catabolic part of BK metabolism [23]. The risks of ACEI-AE are not related to the dose or the specific agent administered (it being a class-adverse reaction) [24][25][26].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241411649
References
- Bernstein, J.A.; Moellman, J. Emerging concepts in the diagnosis and treatment of patients with undifferentiated angioedema. Int. J. Emerg. Med. 2012, 5, 39.
- Mudd, P.A.; Hooker, E.A.; Stolz, U.; Hart, K.W.; Bernstein, J.A.; Moellman, J.J. Emergency department evaluation of patients with angiotensin converting enzyme inhibitor associated angioedema. Am. J. Emerg. Med. 2020, 38, 2596–2601.
- Ferrer, M.; Rodriguez-Garijo, N.; Sabate-Bresco, M. Medical algorithm: Diagnosis and management of histaminergic angioedema. Allergy 2023, 78, 599–602.
- Bernstein, J.A.; Cremonesi, P.; Hoffmann, T.K.; Hollingsworth, J. Angioedema in the emergency department: A practical guide to differential diagnosis and management. Int. J. Emerg. Med. 2017, 10, 15.
- Long, B.J.; Koyfman, A.; Gottlieb, M. Evaluation and Management of Angioedema in the Emergency Department. West. J. Emerg. Med. 2019, 20, 587–600.
- Banerji, A.; Blumenthal, K.G.; Lai, K.H.; Zhou, L. Epidemiology of ACE Inhibitor Angioedema Utilizing a Large Electronic Health Record. J. Allergy Clin. Immunol. Pract. 2017, 5, 744–749.
- Brown, T.; Gonzalez, J.; Monteleone, C. Angiotensin-converting enzyme inhibitor-induced angioedema: A review of the literature. J. Clin. Hypertens. 2017, 19, 1377–1382.
- Straka, B.T.; Ramirez, C.E.; Byrd, J.B.; Stone, E.; Woodard-Grice, A.; Nian, H.; Yu, C.; Banerji, A.; Brown, N.J. Effect of bradykinin receptor antagonism on ACE inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2017, 140, 242–248.e2.
- Beltrami, L.; Zingale, L.C.; Carugo, S.; Cicardi, M. Angiotensin-converting enzyme inhibitor-related angioedema: How to deal with it. Expert. Opin. Drug Saf. 2006, 5, 643–649.
- Aygoren-Pursun, E.; Magerl, M.; Maetzel, A.; Maurer, M. Epidemiology of Bradykinin-mediated angioedema: A systematic investigation of epidemiological studies. Orphanet J. Rare Dis. 2018, 13, 73.
- Pfaue, A.; Schuler, P.J.; Mayer, B.; Hoffmann, T.K.; Greve, J.; Hahn, J. Clinical features of angioedema induced by renin-angiotensin-aldosterone system inhibition: A retrospective analysis of 84 patients. J. Community Hosp. Intern. Med. Perspect. 2019, 9, 453–459.
- Moreau, M.E.; Dubreuil, P.; Molinaro, G.; Chagnon, M.; Muller-Esterl, W.; Lepage, Y.; Marceau, F.; Adam, A. Expression of metallopeptidases and kinin receptors in swine oropharyngeal tissues: Effects of angiotensin I-converting enzyme inhibition and inflammation. J. Pharmacol. Exp. Ther. 2005, 315, 1065–1074.
- Hoover, T.; Lippmann, M.; Grouzmann, E.; Marceau, F.; Herscu, P. Angiotensin converting enzyme inhibitor induced angio-oedema: A review of the pathophysiology and risk factors. Clin. Exp. Allergy 2010, 40, 50–61.
- Kaplan, A.P.; Joseph, K.; Silverberg, M. Pathways for bradykinin formation and inflammatory disease. J. Allergy Clin. Immunol. 2002, 109, 195–209.
- Hebert, J.; Boursiquot, J.N.; Chapdelaine, H.; Laramee, B.; Desjardins, M.; Gagnon, R.; Payette, N.; Lepeshkina, O.; Vincent, M. Bradykinin-induced angioedema in the emergency department. Int. J. Emerg. Med. 2022, 15, 15.
- Gainer, J.V.; Morrow, J.D.; Loveland, A.; King, D.J.; Brown, N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 1998, 339, 1285–1292.
- Pretorius, M.; Rosenbaum, D.; Vaughan, D.E.; Brown, N.J. Angiotensin-converting enzyme inhibition increases human vascular tissue-type plasminogen activator release through endogenous bradykinin. Circulation 2003, 107, 579–585.
- Seyedi, N.; Maruyama, R.; Levi, R. Bradykinin activates a cross-signaling pathway between sensory and adrenergic nerve endings in the heart: A novel mechanism of ischemic norepinephrine release? J. Pharmacol. Exp. Ther. 1999, 290, 656–663.
- Fryer, R.M.; Segreti, J.; Banfor, P.N.; Widomski, D.L.; Backes, B.J.; Lin, C.W.; Ballaron, S.J.; Cox, B.F.; Trevillyan, J.M.; Reinhart, G.A.; et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: Rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–955.
- Adam, A.; Cugno, M.; Molinaro, G.; Perez, M.; Lepage, Y.; Agostoni, A. Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet 2002, 359, 2088–2089.
- Byrd, J.B.; Shreevatsa, A.; Putlur, P.; Foretia, D.; McAlexander, L.; Sinha, T.; Does, M.D.; Brown, N.J. Dipeptidyl peptidase IV deficiency increases susceptibility to angiotensin-converting enzyme inhibitor-induced peritracheal edema. J. Allergy Clin. Immunol. 2007, 120, 403–408.
- Molinaro, G.; Cugno, M.; Perez, M.; Lepage, Y.; Gervais, N.; Agostoni, A.; Adam, A. Angiotensin-converting enzyme inhibitor-associated angioedema is characterized by a slower degradation of des-arginine(9)-bradykinin. J. Pharmacol. Exp. Ther. 2002, 303, 232–237.
- Cugno, M.; Nussberger, J.; Cicardi, M.; Agostoni, A. Bradykinin and the pathophysiology of angioedema. Int. Immunopharmacol. 2003, 3, 311–317.
- Brown, N.J.; Snowden, M.; Griffin, M.R. Recurrent angiotensin-converting enzyme inhibitor--associated angioedema. JAMA 1997, 278, 232–233.
- Sabroe, R.A.; Black, A.K. Angiotensin-converting enzyme (ACE) inhibitors and angio-oedema. Br. J. Dermatol. 1997, 136, 153–158.
- Quan, M. Case study. ACE inhibitor-induced angioedema. Clin. Cornerstone 2009, 9 (Suppl. 3), S34–S35.
This entry is offline, you can click here to edit this entry!