Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Integrative & Complementary Medicine
Omega-3 fatty acids v(ω-3 FAs) such as EPA (eicosapentaenoic acid) and DHA (docosahexaenoic acid) and omega-6 fatty acids (ω-6 FAs) such as linoleic acid and arachidonic acid are important fatty acids responsible for positive effects on human health. The main sources of ω-3 FAs and ω-6 FAs are marine-based products, especially fish oils.
- spectroscopic techniques
- omega-3 fatty acids
- ω-6 FAs
- chemometrics
1. Introduction
Fish oils are good sources of polyunsaturated fatty acids (PUFA) including omega-3 fatty acids (ω-3 FAs) and omega-6 fatty acids (ω-6 FAs). In terms of chemical structures, ω-3 FAs and ω-6 FAs can be differentiated from the first double bond locations, counting from the terminal of methyl groups of the corresponding fatty acid. In ω-6 FAs, the first double bond is located between the sixth and seventh carbon atoms, while for ω-3, the first double bond is located between the third and fourth carbon atoms [1]. Alpha-Linolenic acid or ALA (C18:3, ω-3), eicosapentaenoic acid or EPA (C20:5, ω-3), docosapentaenoic acid or DPA (20:5 n-3), docosahexaenoic acid or DHA (C22:6, ω-3), tetracosapentaenoic acid or TPA (C24:5 n-3), and tetracosahexaenoic acid or THA (C24:6n-3) are among ω-3 FAs, while linoleic acid or LA (C18:2, ω-6) and arachidonic acid or ARA (C20:4, ω-6) are example of ω-6 FAs. The chemical structures of selected ω-3 FAs and ω-6 FAs are depicted in Figure 1.
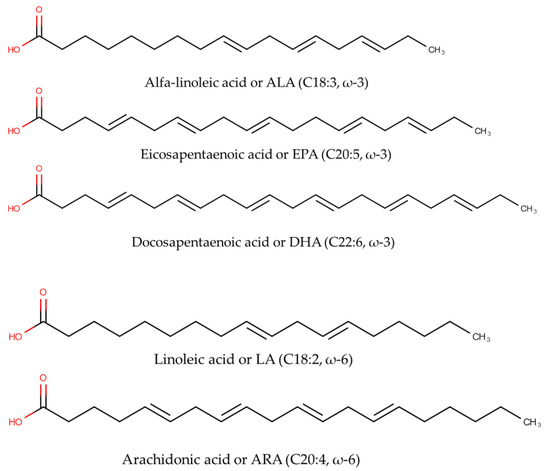
Figure 1. The chemical structures of omega-3 fatty acids including alpha-Linolenic acid (C18:3, ω-3), eicosapentaenoic acid or EPA (C20:5, ω-3), and docosahexaenoic acid or DHA (C22:6, ω-3), as well as omega-6 fatty acids including linoleic acid or LA (C18:2, ω-6) and arachidonic acid or ARA (C20:4, ω-6).
In marine oils, the most dominant ω-FAs are DHA, ARA, and EPA. Linolenic and linoleic acids are essential FAs needed by the human body and contain double bonds that are not synthesized in the human body [2]. ω-3 FAs are believed to contribute positive potentials to human health. The beneficial health effects of ω-3 FAs, especially EPA and DHA, were firstly described in the Greenland Eskimos, who consumed a high seafood diet and exhibited low rates of coronary heart disease, type I diabetes mellitus, asthma, and multiple sclerosis. Since this observation, the advantageous effects of ω-3 FAs have been widely explored. The consumption of foods containing EPA and DHA is associated with low risk of cardiovascular attack [3]. EPA and DHA are essential for proper fetal development and brain development and for lowering depression risk [4]. It is believed that a diet rich in ω-6 FAs could promote the pathogenesis of some chronic inflammatory diseases. These beneficial effects have attracted scientists to explore the natural sources of ω-3 and ω-6 FAs. A well-balanced ω-3/ω-6 FAs ratio is reported to be proven for promoting health and preventing disease formation, mostly in the Mediterranean diet. In addition, the balance of ω-3 and ω-6 FAs is an important determinant in decreasing the risks of coronary heart disease (CHD), cancer, diabetes, arthritis, hypertension, and other neurodegenerative diseases. As a consequence, some research has reported this ratio value. The ratio of ω-6/ω-3 from 3:1 to 4:1 could prevent pathogenesis of various diseases [5].
EPA and DHA cannot be synthesized in the human body de novo; therefore, human and mammals should obtain them from the diet. However, both EPA/DHA and other n-3 long-chain poly-unsaturated fatty acids can be synthesized from their precursor of alpha-linolenic acid (ALA, 18:3 n-3) through the desaturation and elongation of EPA. Unfortunately, the conversion rate of ALA into EPA/DHA is very low because of the limited activity of the ∆6-desaturase enzyme [6] (Figure 2). Some fish species have elongase and desaturase enzymes, which enable them to convert linoleic acid (LA, 18:2 n-6) into ARA and ALA into EPA and DHA [7]. Thus, EPA/DHA must be provided favorably from the diet, especially from the main source of marine-based foods [8]. According to the Food and Agriculture Organization (FAO), the recommended daily intake of EPA + DHA is set between 100 and 250 mg for children up to 10 years, 250 mg for healthy adults, and 300 mg for pregnant women (of which at least 200 mg must be DHA). On the other hand, arachidonic acid, an n-6 polyunsaturated 20-carbon fatty acid formed by the biosynthesis of linoleic acid, was reported to play important roles in infant development [9]. For this reason, the evaluation of ω-FAs is very important to confirm their contents in food and supplement products [10].
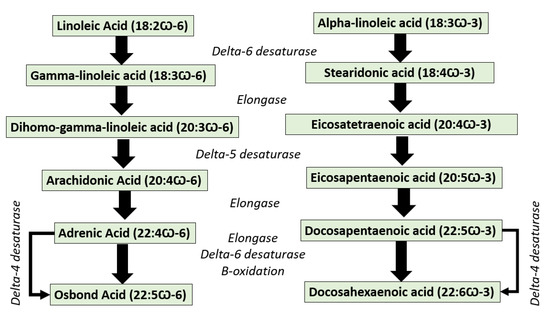
Figure 2. The conversion pathway of omega-3 fatty acids represented by osbond acid and omega-6 fatty acids represented by docosahexaenoic acid.
2. Chemometrics
Chemometrics can be described as the science implementing statistical and mathematical techniques to obtain relevant information related to process, design, and optimal selection by analyzing chemical data [12,13]. In the last two decades, chemometrics has been widely applied in the field of chemistry to enhance analytical methods such as spectroscopy and chromatographic-based methods [14,15]. Chemometrics of exploratory data analysis of Principal Component Analysis (PCA) was applied for authenticating several oils using an open source chemometrics software [16]. Another study has successfully performed the classification of marine fish surimi according to the species, namely white croaker surimi (Argyrosomus argentatus), hairtail surimi (Trichiurus haumela), and red coat surimi (Nemipterus virgatus) [17]. In order to enhance the quality of the authentication purposes, chemometrics of pattern recognition were widely applied. Unsupervised pattern recognition of PCA and supervised pattern recognition of linear discriminant analysis (LDA) and Soft Independent Modeling of Class Analogies (SIMCA) have been generated to investigate the potential substitution of valuable fish species with cheaper materials [18]. A multivariate calibration technique of partial least squares regression (PLSR) has been successfully developed to build prediction models for determining fatty acids content (%) from eight fish oil samples [19]. Figure 3 presents the scheme of chemometrics application for analyzing omega-3 or omega-6 fatty acids using spectroscopic and chromatographic-based methods.
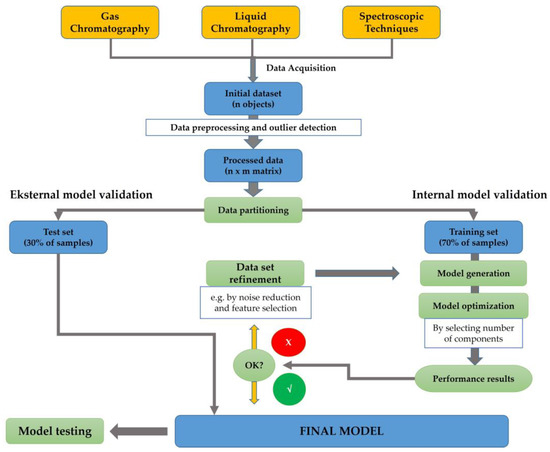
Figure 3. The detailed workflow of analytical methods and chemometrics techniques used for analysis of omega-3 (ω-3) or omega-6 (ω-6) fatty acids using spectroscopic and chromatographic-based methods.
A set of initial data, which are collected from analytical instruments such as spectrophotometer and/or chromatography, could be used for chemometrics analysis. At first, several data pre-processing and outlier detections can be applied to support the subsequent analytical modeling [21]. In the case of supervised pattern recognition modeling, a matrix of data achieved from the pre-processing stage has been divided into two groups consisting of the training set (70% of the data) and test set (30% of the data). This partition will enable the implementation of internal model validation, cross validation, and external validation of the model [22]. The generation of the model followed by the model optimization by components selection are then executed. Performance quality for model accuracy can be evaluated by assessing the determination coefficient of the calibration model (Rcal2) and the determination coefficient of cross-validation (RCV2), whereas the difference or error between the actual and predicted values of the model can be evaluated by root mean square error of calibration (RMSEC) and root mean square error of cross-validation (RMSECV) [23]. It can be noted that the data refinement can be applied to improve the model quality. The final chemometrics model is generated by considering the external validation using test set data. The quality of the prediction should be evaluated using the determination coefficient of validation (Rval2) and root mean square error of calibration (RMSEP). Further, the final chemometrics techniques with appropriate features can be applied for sample evaluation.
3. Official Methods of Analysis of ω-3 FAs and Related Compounds
In the current USP-NF online, three ω-3 FA compounds have been described, i.e., ω-3 free fatty acids [24], ω-3 acid triglycerides [25], and ω-3-acid ethyl esters [26]. Ω-3 free fatty acids are a mixture of ω-3 acids in their free form. They consist mainly of alpha-linolenic acid (C18:3 n-3), moroctic acid (C18:4 n-3), eicosatetraenoic acid (C20:4 n-3), eicosapentaenoic acid (EPA) (C20:5 n-3), heneicosapentaenoic acid (C21:5 n-3), docosapentaenoic acid (C22:5 n-3), and docosahexaenoic acid (DHA) (C22:6 n-3) [24]. Ω-3 acid triglycerides are a mixture of mono, di, and tri-esters of ω-3 acids with glycerol containing mainly tri-esters and obtained either by esterification of concentrated and purified ω-3 acids with glycerol or by transesterification of the ω-3 acid ethyl esters with glycerol [25].
Omega-3-acid ethyl esters are a mixture of ethyl esters, principally the ethyl esters of eicosapentaenoic acid (EPAee) (C20:5 n-3, EE) and docosahexaenoic acid (DHAee) (C22:6 n-3, EE). They may also include ethyl esters of alpha-linolenic acid (C18:3 n-3, EE), moroctic acid (C18:4 n-3, EE), eicosatetraenoic acid (C20:4 n-3, EE), heneicosapentaenoic acid (C21:5 n-3, EE), and docosapentaenoic acid (C22:5 n-3, EE). Tocopherol may be added as an antioxidant [26].
The content of EPA, DHA, and total ω-3 free fatty acids are determined using GC equipped with FID (270°), column: 0.25-mm × 25-m; coated with a 0.20-µm film of phase G16 (Carbowax 20M), injection port: 250°, split ratio: 200/1 temperature programmed: initial: 170° (2 min) then raise to 240° (3°/min, 2.5 min). For verifying the methods, two SSTs (standard solution tests) are used; SST 1 consists of USP ω-3 free fatty acid RS, while SST 2 consists of DHA methyl ester and tetracos-15-enoic acid methyl ester. USP EPA RS and USP DHA RS are used as the standard, and USP methyl tricosanoate RS is used as IS. Detailed preparations of SST solutions and samples can be referred to in this official method [24].
Acceptance criteria: RRt (relative retention time) of alpha-linolenic acid (C18:3 n-3;0.564), moroctic acid (C18:4 n-3; 0.592), eicosatetraenoic acid (C20:4 n-3; 0.776), EPA (C20:5 n-3; 0.793), heneicosapentaenoic acid (C21:5 n-3; 0.887), and docosapentaenoic acid (C22:5 n-3; 0.973) are calculated against Rt (retention time) of DHA (C22:6 n-3; 1.00). Chromatogram of sample should be similar to SST 1. EPA should be NLT 45% and NMT 65%, DHA NLT 10% NMT 30%, and total ω-3 free fatty acids NLT 80%. [24]
Unfortunately, this current USP-NF online (https://online.uspnf.com/uspnf, accessed on 28 May 2023) does not describe the method for determination of similarity of the chromatograms. Alaerts et al. described the application of the correlation coefficient r and the congruence coefficient c for the determination of the similarity [27].
The following official procedure of the general chapter <401> of USP 46-NF 41 may be used for the determination of eicosapentaenoic acid (EPA) (C20:5 n-3), docosahexaenoic acid (DHA) (C22:6 n-3), and total ω-3 FAs obtained from fish, plant, or microbial sources in bulk oils and encapsulated oil, either as triglycerides or as ethyl esters. The term “triglyceride” is applicable to algal oils, fish oils, fish liver oils, and products containing ω-3 FAs in triglyceride form [28]. If using split injection, the GC-FID method is identical to [16], if using splitless injection, the conditions have modified as follows: injection port: 90–250°, column temperature programmed: 90° (2 min) raise to 170° (30/min, 3.0 min), then raise to 240° (3°/min, 2 min). Concentrations of EPA, DHA, and total ω-3 FAs (for triglycerides or ethyl ester) are estimated. Detailed methods including sample preparations, SSTs, and acceptance criteria have been described in this general chapter [28].
The official GC method of the general chapter <401> of USP 46-NF 41 [28] has been applied for the determination of EPA, DHA, and total ω-3 FAs in the monograph of ω-3 acid triglycerides [25], ω-3 acid ethyl esters [23,26], fish oil containing ω-3 FAs [29], and other related monographs including materials from plants. In the current USP-NF online https://online.uspnf.com/uspnf (accessed on 29 May 2023), 15 monographs describe the methods of analysis of ω-3 FAs and related compounds. SSTs and acceptance criteria can be referred to in the respective monographs. Verification or validation methods should be performed prior to routine application of the official methods in the QC laboratory of the pharmaceutical industry. If the results of the SSTs can fulfil the criteria that are described in the respected monographs, there is no need to perform full validation methods [30,31].
4. Chromatographic-Based Methods for Analysis of ω-3 FAs and ω-6 FAs
Due to their capacity as separation techniques, chromatographic-based methods offered excellent analytical methods for analysis of ω-3 FAs and ω-6 FAs in marine products. Currently, the types of chromatograph instruments along with their components, mainly columns and detectors, are available in the laboratories, therefore, the routine analysis of fatty acids can be carried out efficiently and effectively [32]. Chromatographic methods can be used for separation, identification, and quantification of these fatty acids with reliable results. Gas chromatography using detectors such as a flame ionization detector (GC-FID) and mass detector (GC-MS) are the most common chromatographic techniques used for determination of ω-3 FAs and ω-6 FAs. Liquid chromatography tandem with mass spectrometry (LC-MS or LC-MS/MS) is also an effective analytical technique for the analysis of omega fatty acids without the derivatization step [33].
4.1. Gas Chromatography for Analysis of ω-3 FAs and ω-6 FAs
Among analytical methods used for fatty acids analysis, chromatographic-based techniques, mainly gas chromatography, have been extensively reported for analysis of fatty acids including ω-3 FAs and ω-6 FAs. In addition, GC also provides a sensitive, accurate, precise, rapid, and reproducible analysis with efficient separation due to the low mass transfer compared to liquid chromatography.
Gas chromatography (GC) is a chromatographic separation technique based on the difference in the distribution of organic compounds between two non-miscible phases in which the mobile phase is a carrier gas moving through or passing the stationary phase contained in a column. GC is appropriate for analyzing any organic compounds or their derivatives, which are volatilized under the temperatures employed. Mainly, GC is based on the mechanisms of adsorption or mass distribution [34]. The common carrier gas is helium, nitrogen, or hydrogen, and it depends on the column and detector in use. The flow of carrier gas through the column and the detector is performed at a controlled rate or pressure [34,35]. A brief discussion regarding general GC system instrumentation has been previously published [36], while discussion on gas chromatography–mass spectrometry (GC-MS) systems can be referred to in previous published papers [37,38].
Injection may be performed either into a vaporization chamber, which may be equipped with a stream splitter, or directly at the head of the column using a syringe or an injection valve [34]. Today, the most used injectors for capillary GC fall into one of four techniques, i.e., split, splitless, on-column injection, and programmed-temperature vaporizers (PTV) [39]. The liquid stationary phases used in GC are contained in a column that comprises either a capillary column (at least 5 m, 0.1–0.53 mm i.d.) whose stationary phase may be a solid coating on the inner surface (0.1–5.0 µm film) of the column (e.g., macrogol 20,000) or a liquid deposited on the inner surface (e.g., dimethylpolysiloxane); in the latter case, it may be chemically bonded to the inner surface and a column packed (1–3 m, 2–4 mm i.d.) with the stationary phase that may be a solid phase (e.g., alumina, silica) or an inert solid support (usually a porous polymer impregnated or coated with a liquid) [34,35].
Two distinct types of detectors are used in GC, namely mass-dependent or concentration-dependent. A mass-dependent detector responds to mass per unit time entering the detector, not mass per unit volume. Mass-dependent detectors are destructive, and the compounds eluting from the column are chemically modified or destroyed in the detector (e.g., FID, flame photometric detector (FPD), nitrogen chemiluminescence detector (NCD), mass spectrometer (MS)). Concentration-dependent detectors are non-destructive and can therefore be used in series with other detectors (e.g., thermal conductivity detector (TCD), electron capture detector (ECD), Fourier Transform Infrared (FT-IR)) [40]. Detectors for GC can be also classified based on their selectivity. TCD is a universal detector that can respond to all compounds except the carrier gas. Most common GC detectors fall into the selective designation, e.g., FID for all organic compounds that contain C–C and C–H bonds, ECD for organic compounds that contain halogen and phosphorous groups, FPD for S and P compounds, NCD for N compounds, and NPD for pesticides. In addition, MS and FT-IR detectors can be used for identifying organic compounds in samples by comparing their spectra to authentic standards and/or libraries [41].
GC can be applied for qualitative and quantitative analysis. Quantitative analysis can be performed using direct calibration (using external and internal standard methods) and standard addition methods [34,41]. Fingerprint analysis can be performed for the identification and characterization of plant/herbal materials and related products [42]. Identification of organic compounds in herbal or plant materials can be performed using combination GC-MS or GC-HR-MS-MS and many free online MS databases. To have reliable results of analysis, all methods (qualitative and quantitative) should be properly validated prior to routine application. Detailed validation methods for chromatography analysis have been reviewed and discussed by previous publications [43,44].
The advancements in GC instrumentation coupled with data processing have led to the evolution of this method as a reference method for analysis of fatty acids in marine organisms. However, in some cases, the extensive sample preparation including extraction and derivatization steps must be done before being subjected to GC analysis [45]. Fatty acids are non-volatile enough; therefore, the derivatization of fatty acids is required. In GC, the derivatization step is required when the analytes are non-volatile, thermally labile, and low sensitivity. Methylation of FAs is the most common method for derivatization of FAs to provide volatile fatty acid methyl esters (FAMEs). Many methylation techniques are reported in the scientific literature, and among them, acid-catalyzed methylation, alkaline-catalyzed methylation, methylation using boron trifluoride (BF3), methylation with diazomethane, and silylation are the most used [46]. Extraction of fatty acids and other lipid components was typically carried using different methods (direct extraction, maceration, Soxhlet extraction, Folch, Bligh–Dyer) employing organic solvents with different polarities [47].
GC-FID has been developed and validated for quantitative analysis of ω-3 FAs of DHA and EPA in raw and cooked fish [48]. Fatty acids were derivatized as fatty acid methyl esters (FAMEs), and the derivatized fatty acids were subjected to GC-FID analysis. The column used is a capillary column of a GC HP-88 column (60 m length, 0.25 mm ID, 0.2 µm DF) with a polar stationary phase. The validation of GC-FID followed the guidance from the International Conference on Harmonization (ICH) by determining and evaluating some characteristics of performance including linearity with r-value > 0.9995, precision with RSD ≤ 2%, accuracy with recovery percentage of >95%, specificity with good peaks separation, and sensitivity with low detection limits. All validation parameters meet acceptance criteria set by ICH requirements. The developed analysis was successfully employed for determination of EPA and DHA in fish samples. GC-FID is also successful for determination of omega-3 and omega-6 fatty acids including EPA and DHA by applying two different cyanopropyl silicone capillary columns (DB-225ms and DB-23). The method is valid based on ICH requirements [46].
GC-MS, the combination of two powerful instruments, namely GC for separation and MS for detection, has become a popular method for the confirmation and quantitative analysis of ω-3 FAs (EPA/DHA) in supplement capsules containing fish oils. Quantification of EPA/DHA was carried out using selective ion mode (SIM) using m/z 79 as the ion fragment EPA/DHA. Fatty acids were released from triglycerides using KOH (potassium hydroxide)-methanolic solution (followed by esterification with BF3-methanol solution to provide FAMEs). The method was validated according to the ICH (International Council for Harmonization), and all validation parameters fit with acceptance criteria set by ICH requirements. Limit of detection (LoD) values obtained were 0.08 ng and 0.21 ng for EPA and DHA, respectively, with RSD values of intra-day and inter-day variations of <0.59% and 1.00% for EPA and <1.08% and 1.05% for DHA. The percentages of average recoveries for EPA and DHA were 100.50% and 103.83%, respectively. The validated method was successfully used for analysis of EPA and DHA contents in commercial capsules containing fish oils. There is no correlation between the contents of EPA/DHA and the price of supplement capsules [49]. GC-MS was also applied for analysis of ω-3 (ALA, EPA, DHA, DPA) and ω-6 FAs (dihomo-linoleic acid or DGLA and ARA) with valid results [50].
4.2. Liquid Chromatography for Analysis of ω-3 and ω-6 FAs
Traditionally, ω-3 FAs and ω-6 FAs were determined using GC-FID and GC-MS, in which both analytes of ω-3 FAs and ω-6 FAs must be extracted from the marine samples to release them from their triglycerides, and FAs were derivatized to their volatile methyl esters (FAMEs) prior to analysis by GC. Even though GC-FID or GC-MS are the standard methods for analysis of ω-3 FAs and ω-3 FAs, they are very tedious and time-consuming since these methods comprise several steps including extraction, hydrolysis, and derivatization before being subjected to GC-FID/GC-MS. In addition, the use of high temperature during GC measurement can affect the stability of FAs. For this reason, a greener method of analysis based on HPLC, which does not involve the complex step, should be used. Direct HPLC-UV detection is limited for determination of ω-3 FAs and ω-6 FAs since FAs lack chromophore groups requiring low wavelengths, which limits the solvent selection and increases the likelihood of matrix interference. The combination of the dual-gradient HPLC method and charged aerosol detection has enabled the determination of ω-3 FAs and ω-3 FAs in a single analysis without derivatization of analytes as in GC-FID/GC-MS.
-
Liquid chromatography (LC) is a chromatographic separation technique based on the difference in the distribution of species between two non-miscible phases in which the mobile phase is a liquid that eluted through a stationary phase contained in a column. The term LC is synonymous with high-pressure liquid chromatography (HPLC) [35,59]. Particle diameter of the stationary phase for classical HPLC is about 2–10 µm; if the particle diameter is less than 2 µm, the term becomes ultra-high-performance liquid chromatography (UHPLC) [60]. A liquid chromatograph comprises (1) a pumping system, (2) an injector, (3) a chromatographic column (a column temperature controller may be used), and (4) one or more detectors and data acquisition systems [59].
Depending on the type of interaction of the sample molecules with both mobile and stationary phases, different types of interactions can be possible in LC: adsorption chromatography, partition chromatography, ion exchange chromatography, affinity chromatography, and size exclusion chromatography [60]. Stationary phases used for LC are silica or alumina, a variety of chemically modified supports prepared from polymers, silica or porous graphite, resins or polymers with acidic or basic groups, porous silica or polymers, and specially modified stationary phases for the separation of enantiomers (chiral chromatography) [59].
Mobile phases are a solvent or a mixture of solvents, as defined in the individual monograph of the Pharmacopeias. For normal-phase LC, low-polarity organic solvents are generally employed. In reversed-phase LC, aqueous mobile phases, usually with organic solvents and/or modifiers, are employed. Mobile phases may contain other components, for example, a counter-ion for ion-pair chromatography or a chiral selector for chiral chromatography using an achiral stationary phase. The technique of continuously changing the mobile phase’s composition during the chromatographic run is called gradient elution or solvent programming. Mobile phases should be filtered to remove particles greater than 0.45 µm in size (or greater than 0.2 µm when the stationary phase is made of sub-2.0 µm particles and when special detectors, e.g., light-scattering detectors, are used) [35,59].
Many detectors can be used for LC; these include UV-Vis, DAD, fluorescence spectrophotometers, RI, ED, CAD, MS or MS/MS, and MALS [59]. In chemical or pharmaceutical analysis, LC can be applied for identification and purity tests and quantitative analysis (assay) using absolute curve calibration methods and internal standardization methods [61]. Qualitative identification and purity tests of plant/herbal materials and related products using fingerprints have been previously reviewed and discussed [42]. Detailed theory, method development, optimizations, and troubleshooting using LC methods have been described and discussed [60]. Identification of organic compounds or metabolites in plant/herb materials can be performed using combination LC-HR-MS/MS and many online MS databases. Reliable data can be only obtained if all methods used are validated prior to applications; for detailed discussion regarding this matter, refer to the previous publications by Riswanto et al. and Irnawati et al. [14,16]. If the recovery of the method using LC-MS/MS is not acceptable, the matrix effect should be evaluated [62].
HPLC using a charged aerosol detector (CAD) has been used for separation and quantitative analysis of six ω-3 FAs, SDA, EPA, ALA, DHA, DPA, and ETA, five ω-6 FAs, GLA, ARA, LA, adrenic acid, and eicosadienioic acid (EDA), and two omega-9 fatty acids, oleic and erucic acids, in hydrolyzed fish oil-based commercial supplements. CAD is mass sensitive, which can be added to HPLC and other LC system platforms, providing the most consistent response for semi-volatile and non-volatile analytes. This detector works by charging the analytes, and the charged analytes were separated using the column Acclaim™ C30 (250 × 3 mm, 3 µm) at a temperature of 30 °C. The mobile phase used consisted of mobile phase A of water:formic acid:mobile phase B (900:3.6:100 v/v/v) with mobile phase B of acetone:acetonitrile:tetrahydrofuran:formic acid (675:225:100:4 v/v/v/v) delivered in a gradient manner. Figure 4 exhibits the separation profiles of ω-FAs in hydrolyzed fish oil. All ω-3 FAs and ω-6 FAs were well separated with good efficiency and resolution. In addition, the developed method is valid and meets ICH requirements as indicated by acceptable criteria of performance characteristics such as linearity and recovery. LoD values were in the range of 1–10 ng. Using this method, the relative levels of ω-3 FAs, ω-6 FAs, and omega-9 FAs in fish oils were of 92.0%, 3.2%, and 4.5%, with the ratio of ω-3 to ω-6 being 29.0 [73].
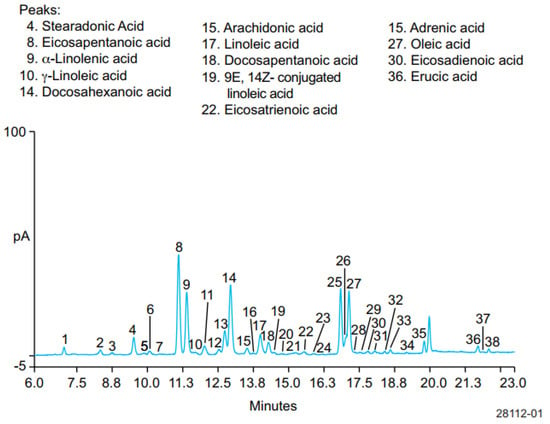
Figure 4. Separation of stearidonic acid (SDA), EPA, ALA, DHA, docosapentaenoic acid or DPA, and Eicosatrienoic acid (ETA); five ω-6 FAs, γ-linolenic acid (GLA), ARA, linoleic acid (LA), adrenic acid, and eicosadienioic acid (EDA); and two omega-9 fatty acids (oleic and erucic acids).
This entry is adapted from the peer-reviewed paper 10.3390/molecules28145524
This entry is offline, you can click here to edit this entry!