Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Neglected Tropical Diseases (NTD) is a group of 20 diseases affecting more than 1 billion people in the world, especially those living in poor communities of tropical areas. Caused by a variety of etiologic agents such as viruses, bacteria, and parasites, these diseases have a great impact on public health, with social and economic consequences to the affected populations.
- covalent target enzyme inhibitors
- sleeping sickness
- Chagas disease
- Malária
1. Introduction
Neglected Tropical Diseases (NTD) is a group of 20 diseases affecting more than 1 billion people in the world, especially those living in poor communities of tropical areas. Caused by a variety of etiologic agents such as viruses, bacteria, and parasites, these diseases have a great impact on public health, with social and economic consequences to the affected populations [1]. These diseases are often carried by animal reservoirs, some of which are difficult to control. By affecting impoverished populations with little to no access to public health policies, NTDs do not attract enough interest for the pharmaceutical sector, due to the lack of financial profit. As a result, the chemotherapeutic arsenal is scarce for the majority of those diseases, and the lack of an efficient treatment leads to more than 170 million deaths per year [1]. Although this scenario has gradually improved, the investments keep going below the necessity. Research for new chemotherapeutic alternatives for these endemic health situations must be highly emphasized.
Surprisingly, new studies on covalent inhibitors and targeted covalent inhibitors (TCI) comprehend a potential new class of molecules with improved efficacy and security for NTDs. Compared to reversible inhibitors or mechanism-based inactivators, TCIs exhibit unique selectivity profiles. Additionally, while these previous methods are mainly limited to enzymes, the TCI approach is more versatile and can be employed for a wide range of druggable proteins [2].
TCIs can create a stable covalent bond with the target protein active site by positioning their moderately reactive electrophile warhead adjacent to a particular nucleophile amino acid residue, usually cysteine. Their pharmacological impact is sustained, while the unbound drug is quickly and continuously removed from the body from a pharmacokinetic perspective, which makes the latter less toxic [3][4]. TCIs have been employed for the development of new molecules for these three diseases over the past few years, leading to promising results and new alternatives for their treatment.
It is important to notice that the complex TCI-target is formed only after a molecular recognition of the ligand by its target molecule. TCIs are developed to not only have a reactive warhead to create a covalent bond but also must bear structural features for selective and specific interactions with its target; otherwise, no good inhibition is obtained [3].
2. Covalent Inhibitors for Neglected Diseases
Numerous covalent inhibitors and their corresponding warheads are examined. On that matter, Ki, k2nd, kinact are some of the parameters discussed and will be briefly introduced here. It is important to stress that covalent inhibition is a two-step procedure as pictured in Scheme 1. Some of the constants involved in the process are described below.

Scheme 1. Simplified scheme showing the formation of a covalent complex and its constants. P-I is the complex in which P and I form non-covalent interactions between them. P-I* consists of covalent binding between the two molecules.
kinact is the rate constant defining the velocity at which the enzyme is being inactivated, being measured in s−1. Ki is defined as the concentration of inhibitors responsible for half of the maximum rate of the enzyme inhibitory activity, which is closed linked to binding affinity. k2nd is a constant frequently recommended to be used in covalent inhibitors studies but not usually applied [5]; It corresponds to kinact/Ki.
This constant kinact shows the rate at which the covalent bond is formed, primarily when analyzing irreversible inhibitors, and is a pseudo-first-order rate constant defining the rate of enzyme inactivation at full target occupancy [5]. Despite being a good way to understand the nature of the inhibitor, many studies correlating SAR with IC50 have over-relied on IC50, and, as recommended by Strelow, these studies “should be restricted to preliminary investigations” [5].
The correlation between kinact/Ki and IC50 improves as kinact decreases, and the covalent term of the equation becomes less significant. To go further into the details about covalent inhibition mechanisms, see reference [5].
2.1. Covalent Inhibitors Studies on Human African Trypanosomiasis (HAT)
The actual therapy for HAT is based on a few available drugs: pentamidine and suramin for first-stage HAT, while second-stage HAT is treated with eflornithine, melarsoprol, and nifurtimox (Nfx); Currently, as no efficient vaccine is available, treatments have been focused on combination therapies, but, still, several adverse effects have been observed, such as melarsoprol high neurotoxicity [6].
2.1.1. Oxidizing Cycle
One of the most known pathways studied against HAT is the parasite oxidizing cycle since oxidative stress plays a major role in parasite survival [7][8]. Figure 1 shows this pathway; components shown in red are the ones currently being targeted for covalent inhibition explored.

Trypanothione Reductase (TR)
Trypanothione reductase is the most thoroughly studied enzyme of the trypanothione metabolism, as it is not expressed in mammalian hosts, and many of its inhibitors have been identified. As the parasite does not have a glutathione reductase system, which is essential for cellular redox homeostasis [11], the only structure that connects NADPH and thiol-based redox systems is TR [12].
TR is an oxidoreductase that catalyzes NADPH-dependent reduction of trypanothione (bis(glutathionyl)spermidine) disulfide TS2 to trypanothione T(SH)2 (Scheme 2). Inhibition of this enzyme makes the parasite susceptible to oxidative stress and cellular damage.

Scheme 2. TS2 reduction reaction.
Lu et al. studied ebselen (1) (Scheme 2), a benzisothiazolinone derivative, anti-oxidative, and anti-inflammatory seleno-organic compound, found through high-throughput screening (HTS) for TR inhibition [13]. It presented an interesting antibacterial activity as a competitive inhibitor of bacterial tryparedoxin. They consequently examined the action of its analog, ebsulfur (2), against trypanosomes and TR. The results showed this molecule is active against T. brucei, as seen in Figure 2, with a IC50 of 0.092 μM in one of the strains, and showed a time-dependent inhibition of TR, indicating covalent inhibition. The inhibition mechanism discussed by the authors focuses on the thioether group present in the molecule, which covalently binds to a cysteine residue in TR.

Figure 2. (a) Main compounds studied for HAT treatment by covalent inhibition and parameters observed; (b) Protein interaction with the molecule 6. Adapted from [14].
In another approach involving TR inhibitors, Marcu et al. focused on the possible effects of chloroacetamide derivatives on inhibition of T. brucei TR, expanding the SAR study to tricyclic heterocycles using phenothiazine and phenoxazine [15]. Despite the lack of data, the authors claimed that compounds 3 and 4 were remarkable time-dependent inhibitors, and halogens present in their structure suggest a possible nucleophilic substitution mechanism. For more details about TR inhibition and related molecular mechanisms, read the review written by Tiwari et al. [16].
Trypanothione Synthetase (TryS)
Trypanothione synthetase catalyzes the synthesis of N1,N8-bis(glutathionyl)spermidine (trypanothione), the main low molecular thiol that supports many redox functionalities in trypanosomatids.
Although TryS was proved to be druggable, the design of inhibitors has been challenging due to specific characteristics of this enzyme. TryS active site accommodates four substrates and contains several regions involved in substrate binding that are highly dynamic [14].
To explore TryS inhibition, Benítez and colleagues utilized ebselen (1), a previously identified multi-TryS molecule [9]. The ENAMINE library(with a true-hit rate of 0.056% and 51,624 compounds) screening identified select hits that showed significant specificity against TryS. However, only ebselen (1) displayed a balanced antitrypanosomal and TryS inhibitory activity. The researchers conducted experiments to determine its mode of action and confirmed its covalent inhibition through the formation of Se-Cys (S) bonds, primarily through MS/MS analysis.
Tryparedoxin
Another step of the oxidizing cycle is catalyzed by tryparedoxin (Tpx). This enzyme is a distant member of the human thioredoxin family, and, along with T(SH)2, catalyzes the synthesis of compounds required for DNA synthesis, making it important for parasite replication.
A HTS investigation by Fueler et al. led to 36 compounds that inhibited the peroxiding system by ≥20% at a concentration of 40 μM [10]. As the following experiments suggested, these authors proved Tpx to be an in vivo and in vitro target and characterized compounds 6, 7 and 8 (Figure 2) as time-dependent ones through a MAL-PEG-based gel shift assay. It was also observed that the presence of the chloromethyl substituent was essential for covalent binding. Tpx Cys40 likely attacks the electrophilic carbon linked to the chlorine groups in a nucleophilic substitution, facilitated by some non-covalent interactions, especially hydrogen bonds (Figure 2b). A detailed study of the mechanism of these types of molecules was then performed by [17].
2.1.2. Rhodesain (RD)
Rhodesain is an essential protein for parasites crossing the blood brain barrier (BBB), reaching the central nervous system. It is a key cysteine protease for the survival and infectiousness of T. brucei rhodesiense. It is also predominant in lysosomes, where it exerts its protease activity. RD has demonstrated to be a validated target because of its significance for the survival of Trypanosoma sp., and several efforts, especially those linked to covalent inhibitors, have been made in the last 10 years [18][19].
Schechter and Berger nomenclature towards sites is present in the Figure 3. Combinatorial chemistry throughout the years were applied to synthesize compounds prone to bind to the sites in several papains. That way, similar trends in cruzain and rhodesain are going to be seen, for example [20].

Figure 3. The nomenclature regarding papain-like proteins according to Schechter and Berger [20].
Peptidic and Pseudopeptide Compounds
Kerr et al. broadened the SAR research of peptidyl vinyl Michael acceptor derivatives for RD inhibition by synthesizing a series of peptide-based compounds with the vinyl sulfones, such as K11777 [18], as lead compounds considering their structures cocrystallized with RD available in PDB [18][19]. Compound 9 presented Kinact of 0.00255 ± 0.00075 min−1, and a high k2nd of 67,000 ± 432 × 103 M−1 min−1, with docking studies suggesting many hydrogen bonds with the active site (Figure 4).

In a continuation of these endeavors, Ettari et al. exploited lead compound 9, obtaining two potent inhibitors with submicromolar EC50 against the parasite [25]. Based on Ki and k2nd values, they concluded that 3,5-difluoro and 4-CF3 phenyl moieties were important in P3, the residue in the side of peptide in the N-terminal moving further to the cleavage point (Figure 4). However, no direct association was found between enzyme activity and antitrypanosomal activity. In this context, they hypothesized that the divergence was related to enzymatic hydrolysis susceptibility and that better stability might be reached by stereochemistry alterations.
In the following year, the same group attempted to enhance the stability properties of previous compounds through the incorporation of halogens and cyclohexane in P3 and P2, the aminoacid residue of the peptide in the N-terminal moving further to the cleavage point respectively [26]. As a result, two compounds were shown to be interesting hits, with highlight being 14 (Figure 4) of k2nd 52,200 ± 26,200 M−1 min−1 and Kinact of 0.0006 ± 0.0001 min−1, which balanced interesting antitrypanosomal and inhibitory activity.
The presence of bulkier groups in P1 residue, the immediate N-terminal to the cleavage site, was discouraged for further experiments as it demonstrated lack of affinity towards RD in P1′, the immediate C-terminal to the cleavage site. This led Previti et al. to enlarge the SAR and perform alterations in P3, especially [27]. In P1 and P2, hPhe and Phe were maintained, respectively, as they showed good activity, and a variety of groups were tested in P3. The peptide bond was substituted by a urea bond, an amide bioisoster, to provide more stability. The compound that presented the best inhibitory activity towards RD did not have good antitrypanosomal activity. These results suggested that aromatic substituents in P3 were essential to modulate properties concerning cell permeability. The compound which best balanced antitrypanosomal and RD inhibition activity was 15 (Figure 5), with a Ki of 0.51 ± 0.36 nM and EC50 of 1.65 ± 0.07 μM. The presence of chlorine and methyl probably improved cell permeability, thus reaching interesting antitrypanosomal activity.

Figure 5. (a) Docking experiments with 19 and RD. In this figure, hydrogen bonds between several residues and 11 can be observed, and possibly facilitate the nucleophilic action of Cys25. Adapted from [26]; (b) Docking experiments carried out by Ettari et al. with 11 and RD [25]; (c) Docking experiments carried out by Previti et al. with 15 and RD [27]; (d) 14 and RD, active site [28].
Most recently, Previti et al. performed docking experiments with 10 (Figure 5) and hypothesized that the carbonyl in P3 did not perform any major non-covalent interactions, leading to investigation of alternative substituents such as amines and a vinyl ester warhead in P1 [28]. As a result, they obtained 3 interesting hits towards RD, 12, 13 and 14. The presence of vinyl ester instead of vinyl ketone in P3 resulted in a loss of affinity (10, 1.1 pM → 12, 5.8 nM). Compound 14 presented the best antitrypanosomal activity, EC50 = 7.0 ± 1 μM, and the docking experiment (Figure 5) revealed its possible mode of interaction.
On the other hand, Chio et al. [29] kept the homophenylalanine (hPhe) at the P1 site unchanged, as it has been shown to be a critical structural requirement for affinity towards the target enzyme. Following the methodology of a previous study [30], they designed a series of aza-nitrile compounds [31] where Phe and Leu residues, typically preferred by rhodesain, were sampled at the P2 position. The amino group of the P2 substituent was protected with a fluoro-benzoyl group, which spanned into the P3 region to optimize interactions with the S3 pocket. All the compounds showed activity in the range of 16–122 nM. Compound 13 and 14 were the most promising, as the former showed interesting antitrypanosomal activity, likely due to its high lipophilicity compared to other compounds in the series, and the latter displayed the best activity against rhodesain, as well as interesting antitrypanosomal activity (Figure 4). These compounds represent new lead compounds for further investigation.
In another approach, Ettari et al. designed RD inhibitors containing a 3-bromoisoxaline moiety as a warhead and a peptidomimetic scaffold as a recognition moiety towards RD [32]. Compounds with interesting activity were obtained from the lead compounds 17 and 18, highlighting the compound 19 with Ki (RD) of 0.96 μM, inferring that 3-bromoisoxaline might be a good warhead when coupled with an interesting recognition motif (Figure 6).
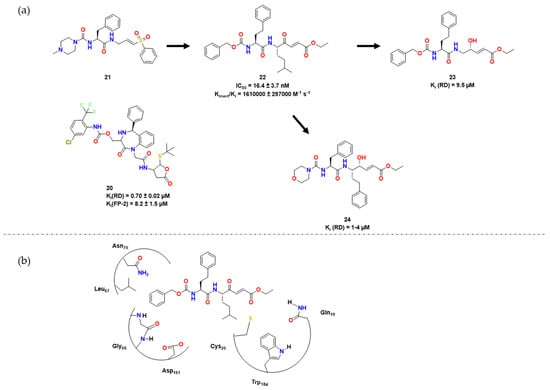
Figure 6. (a) Leads synthesized in [32][33] (b) Docking experiment with compound 22 and RD (adapted from https://www.zotero.org/google-docs/?xzROTL (accessed on 18 March 2023) [32][33]).
Following the work of [32], Chio et al. synthesized a series of compounds containing a vinyl ester as a warhead in place of 3-bromoisoxaline [30]. Among the compounds, 20 (Figure 6) was the most active against both RD and the parasite, providing a good lead for further work. Because of its high lipophilicity, this compound was hypothesized to cross biological membranes more effectively, which possibly contributed to its high activity.
Also in a continuation of the study developed in [32], Royo et al. used 21 as a lead compound and envisioned similar dipeptidyl enoate inhibitors bearing a carbonyl group instead of a sulfonyl one and an alpha carbon group in P1 [33]. Compound 22 was then obtained and shown to be a potent inhibitor of RD, with IC50 of 16.4 ± 3.7 nM and kinact/Ki of 1,610,000 ± 297,000 min−1μM−1. The researchers performed docking experiments to confirm that compounds 21 and 22 had a similar conformation, in which their warheads are positioned close to Cys25. The ester group of 22 possibly interacted with Gln19, Trp184, and His162, similar to the sulfone group of 21. The researchers then tested these dipeptidyl enoates against other cysteine proteases besides RD, such as falcipain and cruzain. Their goal was to optimize interactions at S1, S2, S3, and S1′ [34]. Compound 24 showed the best inhibitory activity against T. brucei. In vitro assays revealed that the IC50 values of 22 and 24 varied between 0.3 and 9.5 μM. Compound 24 was also active against P. falciparum, with an IC50 of 6.81 μM [34] (Figure 7).

Using a different approach, Ettari et al. tackled the high cLogP and molecular weight from previous leads, especially 25, and synthesized one compound in the low micromolar range activity, 26 [36], being interesting also due to its minor cytotoxicity towards mammalian cells (TC50 > 100 μM), besides presenting an IC50 of 5.3 ± 0.03 µM. Concomitantly, the same group explored the benzodiazepine scaffold linked to bromoisoxaline as a warhead, as employed by [32], maintaining the adamantyl group (Figure 7) [35]. They could then keep activity (27, Ki = 1.26 μM, Figure 8) similar to the lead compound and assert the importance of the adamantyl group for the modulation of physicochemical characteristics and, consequently, for trespassing the parasite membrane [35].

In order to optimize compound 21 (Figure 8) [38] and to improve RD inhibition and selectivity toward cathepsins L and B, Jung et al. explored SAR changing the recognition moiety and substitution pattern near the warhead [39]. As a result, aromatic insertions on P3 were interesting for inhibition against RD (30, Ki = 12 nM). As for the selectivity, the incorporation of 4-methyl group in the phenyl ring improved selectivity in comparison with human cathepsins (37, Ki = 8 nM, 12× CatL and 200× CatB). Also, it was noted that 29 showed interesting potency towards RD with a dissociation constant of the initial encounter complex Ki = 24 nM, and with high affinity complex formed in the second step (Ki* = 3 nM) (as Jung et al. defines it, “Ki* denotes the dissociation constant of the high-affinity complex in the case of biphasic, time-dependent inhibition”). Docking studies also suggested this complex to be stable (Figure 9).
As for the last highlight, they compared compounds 28 and 29 as they only differ by the presence of fluor (Figure 8). For its absence, 29 proved to be an irreversible covalent inhibitor.
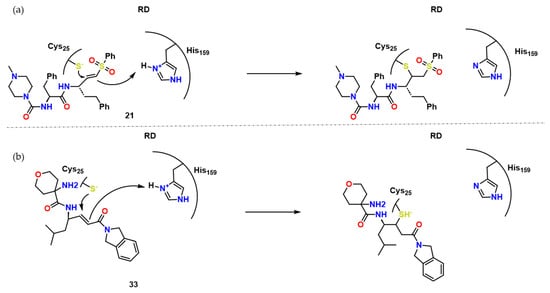
Finally, Chenna et al. described a series of reversible time-dependent inhibitors of cruzain-based on compound 21 and GSK2793660 (compound 33) [21]. These two compounds, especially due to its ability to form irreversible covalent bonds (Figure 10), presented toxicity issues. These compounds, 21 and 33, led the authors to synthesize compounds with less electrophilic moieties, such as 2-vinyl-pyrimidine with Ki* = 0.1–0.4 μM. Soon after, docking studies suggested that heterocycle inhibitors might perform Michael reversible addition to Cys25, such as the example shown for compound 29 in Figure 11.

Figure 10. Compound 34 and its possible interaction towards RD. Adapted from [21].

Figure 11. Non-peptidic compounds synthesized by McShan et al. [40].
Among the hit compounds, 31 presented an interesting EC50 in T. brucei of 5.8 μM, similar to what was pointed out from 24 previous kinetics studies. Compound 32 also deserved a highlight due to its values of EC50 being widely correlated with Ki*, what is a good indication of the correlation of activity with covalent inhibition (Figure 8) [21].
Non-Peptidic Compounds
In recent years, RD inhibition has been also explored with non-peptidic compounds as an attempt to improve pharmacokinetics characteristics in comparison to what was obtained with peptidic compounds [40].
McShan et al. used the irreversible tethering method to explore a library of 200 electrophilic fragments that could potentially lead to covalent inhibitors [40]. In contrast to peptidic ones, compounds 34 and 35 were found to be non-peptidic covalent inhibitors of papain and might possess interesting pharmacokinetic properties. Additionally, these substances had low cytotoxicity and low micromolar activity against T. brucei (Figure 11). The authors also stated a possible mechanism of interaction between the covalent fragments studied and the target [41], which is depicted in Figure 12.

Figure 12. Mechanism of action concerning the covalent fragments studied by McShan et al. [40].
In another approach, Ehmke et al. explored the SAR of aryl nitriles as covalent inhibitors of RD and hCatL, human analogous to RD [41]. They hypothesized that the toxicity of the compounds was directly associated with their warhead electrophilicity and used DFT calculations to understand why some electrophilic moieties had higher inactivation rates than others. Finally, two compounds (38 and 39) with a single digit nanomolar activity were obtained towards RD (9 nM and 2 nM, respectively), showing low selectivity towards the human analogue.
In an attempt to improve the selectivity of these both compounds, Ehmke et al. also constructed another series of compounds, varying P1, P2 and P3 (moieties linked to S1, S2 and S3 as shown in Figure 13) [22]. One of the key findings of the study was that variations in the S1 region did not have a direct impact on inhibitory potency. However, the S2 region was found to be the primary contributor to binding affinity. In particular, compounds with cyclohexyl-based substituents, such as compound 39 shown in Figure 13, showed the most promise. The study also revealed that the S3 region preferred aromatic vectors, likely due to π-stacking interactions. Compounds 38 and 39, with Ki (RD) = 3 nM, were found to be the best candidates when varying the S3 region. Of the two, compound 39 demonstrated greater drug potential due to its lower activity against hCatL (Ki = 32 nM), indicating better selectivity.

In a continuation of studies from [22] and observing the little influence P1 had on activity and affinity towards rhodesain, Schirmeister et al. published a literature review on cathepsin L inhibitors in an attempt to expand the SAR of RD inhibitors focusing in the S2 and S3 regions (Figure 14) [38].

Figure 14. Adapted from [38] Compound 41 and its interactions with RD obtained from docking experiments. There is no interaction of thioimidate with the target according to the docking experiment.
The preference of RD for hydrophobic residues was corroborated with 3- and 4-Br-Phe residues presenting interesting activities. The authors also noted that the compound with a triaryl extremity showed interesting inhibition against RD (41, Figure 13, Ki = 5.3 nM) and a selectivity index of 200 versus cathepsin L.
Crystallographic experiments were also performed in addition with compound 41. These led to the discovery of some hydrophobic interactions next to S2, similar to what was previously proposed [22] (Figure 13), hydrogen bonds, such as between the ligand and Gly66 backbone, and the covalent bond between sulfur from Cys25 and the warhead (Figure 15).

Although compound 41 inhibitory activity against the enzyme was at nanomolar range, the same was not translated into antitrypanosomal activity shown by ATPlite assay (EC50 = 8 μM), being less significant than what was observed previously [22]. One possible explanation was the lack of basic groups in 41, shown to be a good characteristic of those compounds that presented better antitrypanosomal activity [22][38].
Using a library of 49 hCatL inhibitors, Giroud et al. performed a phenotypic screen to identify potential hits against RD. In this context, a macrocyclic lactam presented an activity of 11 nM in Ki for RD (compound 42, Figure 15) [42]. Soon after, the same group simplified the molecule and obtained two compounds with similar activities (43 and 44) at 7.4 nM and 93 nM, respectively [23]. Even though the activity was lower than in previous studies, these compounds were more selective against hCatL. While the first study pointed out a Ki of 10 nM for this enzyme, compounds 43 and 44 presented 0.6 and 0.2 nM, respectively, showing a good potential for this kind of structure.Compound 45 which remarkably inhibited cell growth of T. b. rhodesiense (IC50 (48 h) = 64 nM) and had its structure evaluated towards RD (Figure 16).

Figure 16. Compound 45 and its interactions towards RD [23], highlighting the intramolecular hydrogen bond.
2.1.3. Other Compounds and Targets Related to HAT
Pterine Reductase 1
Tetrahydrobiopterin [H4B] is a necessary cofactor for enzymes such as aromatic amino acid hydroxylases to catalyze different hydroxylation processes. H4B can be generated from GTP or rescued from dihydrobiopterin [H2B] by NADPH-dependent dihydrofolate reductase in humans or NAD[P]H-dependent 6,7-dihydropteridine reductase which regenerates quinonoid dihydrobiopterin, a byproduct of hydroxylation processes. Biological and genetic evidence suggests that trypanosomatids lack the ability to synthesize pterins de novo, and are totally dependent on the uptake of extracellular pterins for growth [43]. Pterin reductase 1 [PTR1] is an enzyme that reduces dihydrobiopterin in Trypanosoma spp., being essential for its survival.
Over the past 10 years, papers have been published, in which pyrimidine-based compounds have been exploited for protein kinase inhibition, topoisomerase inhibition, antibacterial, anti-inflammatory and antiparasitic activity, and dipeptidyl peptidase IV inhibition. Besides, pyrrolopyrimidines had the advantage of carrying a pharmacophore similar to the recognition motif of the parasite P2 aminopurine transporter.
Khalaf et al. considered 2,4-diaminopyrimidines to be important substructures at the outset of this work [44]. Recognizing that physicochemical properties also play an important role in the biological activity of substituted pyrimidines, they investigated 4-alkoxy and 4-alkylamino substituents. Also, this requirement for transport into trypanosomes and the possibility that the specific transporters prefer 4-aminopyridine to engage the hydrophobic pockets of PTR1 evident from crystallographic studies, 5-alkyl, 5-aryl, 6-alkyl, and 6-aryl pyrrolopyrimidines together with 5,6-disubstituted compounds were all studied (Figure 17a). Even though compounds 46 and 47 did not exhibit the best enzyme and in vitro activity, they might be investigated further for the inclusion of a formaldehyde motif, which could potentially form a reversible covalent bond to Cys168.

Figure 17. (a) Hit compounds with interesting activity towards pteridine reductase; (b) Hit compounds with interesting activity towards CLK1, adapted from [45]; (c) ligand interaction site taken from Crystal structure of CLK1 and 49. Adapted from [46]; (d) Compound 50—covalent inhibitor of CatL; (e) Hits compounds from [47].
Kinase CLK1
CLK1 is a crucial protein for mitosis in Trypanosoma spp., and is a target for the antibiotic hypothemycin, highlighting its potential as a drug target [48]. In their research, Saldivia and colleagues found a link between the inhibition of recombinant TbCLK1 and the death of the parasite [45]. Only compounds containing Michael acceptors were studied, leading to the identification of compound 48. To establish the significance of the Michael acceptor for inhibition activity, they synthesized 49 without the unsaturated bond, which in turn abolished compound activity (Figure 17b). Crystallographic analysis revealed that Cys215 forms a covalent bond with the Michael acceptor, while Asp218 appears to be involved in an ionic bond with an amine. Additionally, the nitrogen from Pro214 is close to the benzyl linked to pyrrole, and Tyr212 appears to form hydrogen bonds with oxygen through its backbone (Figure 17c).
Cathepsin-like (TbCATL)
Two cathepsin-like proteins are of paramount importance inside the parasite: TbCATB and TbCATL. In recent studies, however, only TbCATL showed to be essential for the parasite [37]. Compound 50 selectively inhibited the activity of TbCATL in trypanosomes without affecting TbCATB. This molecule showed a dose-dependent inhibitory effect with an (IC50) of 0.39 µM, indicating prominent inhibition of TbCATL (Figure 18d). Steverding et al., lastly, stated that TbCATL is essential for the survival of bloodstream forms of T. brucei and this suggests that future drug development programs shall focus on the rational design of TbCATL inhibitors [37].

Glyceraldehyde-3-Phosphate Dehydrogenase [GAPDH] and TR
Considering the design of new drugs, molecular hybridization has been gaining importance in the last few years. Bellutti et al. aimed to modulate two vital targets in T. brucei and T. cruzi, TR and GAPDH [47]. For that, they considered two different lead compounds and synthesized a hybrid, leading to 10 different derivatives (One of the lead is compound 51, Figure 18e). An interesting feature present is the amine, holding a positive charge at physiological pH, which would present interaction with a pocket filled with negative residues. Compound 53 covalently inhibits the GAPDH through Cys166 and presented the best activity against the two enzymes with an IC50 (GAPDH) of 5.3 μM and Ki (TR) of 2.32 μM. It is worth mentioning though that this trend was not corroborated by in vivo activity, being one of the weakest inhibitors of the series [50] the best one considering in vivo activity, which would suggest off-target activity.
2.1.4. Natural Compounds and Derivatives
The natural world is certainly a good source to start searching for new possible drug candidates [49]. In an isolated study, Oli et al. took advantage of the great structural variety of marine sponges and the fact that some of their extracts were tested against cysteine proteases [50]. From bioactivity-guided fractionation of the crude cyclohexane extract, they obtained plakortide E [51] and found out it had a IC50 of 5 μM in T. brucei and a time dependent IC50 towards RD, varying from 257 μM in 5 min to 77 μM in 60 min, which is indicative for a covalent inhibition given the presence of Michael acceptor.
Considering various compounds previously reported for trypanocidal activity, as well as the recent discovery of furanoheliangolides with high antitrypanosomal activity, the interest in such compounds has increased indeed. Lenz et al. showed that the natural 4,15-isoatriplicolide type sesquiterpene lactones inhibited trypanothione reductase; particularly, 4,15-isoatriplicolide tiglate exhibited exceptionally potent activity against T. brucei rhodesiense with an IC50 value of 15 nM [46]. In 2019, this same group synthesized compound 55, which also proved to be an interesting compound, presenting TbTR inhibitory activity of more than 10% in only 15 min.
Zhang et al., on the other hand, approached natural phenyl vinyl sulfone compounds-derivatives [24]. They observed compound 56 to be inactive against RD at 20 µM after 1 h incubation, Kinact/Ki of 99 M−1 s−1 and to show reasonable in vitro activity, with an IC50 of 5.97 µM. Another interesting feature was its inactivity towards human analogous cathepsin L. Cys25 was discovered to be adjacent to the vinyl moiety in docking investigations. The homophenyl is near the P1 site, while the quinoline is next to P2. The phenyl moiety was predicted to interact with Gln19 and His162, while quinolyl had hydrophobic interactions with Met68. On the other hand, P3 did not have a moiety of its own. Thus, regarding these experiments, it is of interest to highlight the synthesis of compound 57, which presented interesting in vivo antitrypanosomal activity, and 58, which presented a great selectivity index of over 159 (Figure 19).

Figure 19. Distribution of pockets that will be used in this part of review. Also, it is noted that the parts of molecule which are nearest to these pockets are P1, P2, and P3. pdb: 4QH6.
2.2. Covalent Inhibitors Studies on Chagas Disease [CD]
2.2.1. Cruzain [CZ]
Cruzain is the largest cysteine protease from T. cruzi, and it is associated with many crucial biological processes inside the parasite, such as differentiation, invasion, and proliferation in host cells [52]. The sites most explored regarding drug discovery and this enzyme are S1, S2, and S3, as shown in Figure 19.
Peptidomimetics
One of the major efforts in assessing drug design against CD and CZ inhibition involves peptidomimetic compounds. The use of these structures comes from the fact that cysteine proteases target protein-like structures. CZ, as a cysteine protease per se, is not different, and using this scaffold as a lead has allowed researchers to design many potential inhibitors [53].
Avelar et al. used a molecule established by [54] as a lead (59, Figure 20), and explored nitrile as a warhead for TCI against CZ [55]. After many modifications in P1, P2 and P3 (as seen in Figure 18), they synthesized a derivative containing 1-methyl-3-tert-butyl pyrazole in P3 and chlorine in meta for P2 (compound 63, pKi = 7.2). Furthermore, crystallographic experiments revealed that phenylalanine did not penetrate as effectively as m-chlorine in pocket S2. The authors determined that efficient penetration of S2 was only achievable when 3-chloro and 1-methyl-3-pyrazole groups were present. Despite finding this information, they were unable to demonstrate a substantial structure-activity link for amastigote forms.

Compounds 61 and 62 were the two most active compounds against CZ synthesized by the aforementioned group, with pKi equal to 7.8 ± 0.03 and 7.6 ± 0.02, respectively (Figure 20).
The mechanism of action concerning the molecules cited is thoroughly explored by Dos Santos et al. [58]. Experimental data has pointed out the covalent, reversible inhibition of nitrile-based compounds, depending on Cys25 and His162 residues. The imidazole group in His162 polarizes the SH group of Cys25 and enables its deprotonation, promoting Michael substitution on the ligand (Figure 21).

Figure 21. Proposed mechanism of covalent inhibition concerning nitrile compounds such as the ones shown in Figure 19 [58].
Through further efforts related to Avelar et al. [55], Lameiro et al. attempted to optimize compound 60 to achieve metabolic stability by substituting amide for trifluoro groups [53]. The authors also explored the exchange of Phe by Leu at P2, the stereochemistry of adjacent CF3 carbon, and how the bioisosteric replacement H/F affected activity, besides exploring selectivity towards various Cysteine proteases.
On the other hand, Cianni et al. used target-based molecular design to construct a series of potential CZ inhibitors based on compound 58 obtained by Avelar et al. [56]. They then hypothesized that Asp16, Gly66 (S2), and His162 (S1) were critical amino acids for CZ inhibition based on PDB data (PDB code: 1ME3). Furthermore, S3 had not been thoroughly explored before their work. In this regard, they used compound 58 [55] as a prototype and searched for it in silico, kinetics, and thermodynamics experiments to evaluate many derived analogs. The researchers determined that Ser61 interacted better with chlorine atoms in meta, which might explain halogen interactions observed through molecular dynamics. To further understand this pattern, the authors examined different halogens in meta position and discovered that fluorine allowed for a more violent rotation of the molecule and proposed a matched pair molecular analysis, focusing on modifications in P3. The most significant change observed was from m-H to m-I, followed by m-H to m-Cl, as corroborated by molecular modeling experiments. In addition, they mentioned bond donors were less potent than -Cl, which indicates the existence of halogen bonds.
The m-chlorophenyl, m-bromophenyl, and m-hydroxyphenyl moieties presented the correct orientation inside P3 to interact with the oxygen of the Ser61 residue, according to molecular dynamics studies. To properly examine this pattern, they synthesized 11 novel CZ inhibitors with various substituents in P3. The results were in good agreement with the respective molecular dynamics simulations, once the best compounds were precisely those with a lower fluctuation of the P3 ring [55] (Figure 22). The substitution of a hydrogen atom in meta position in P3 for an iodine atom increased the pKi by 0.8 log units.

Alves et al., following Avelar et al. study [55], verified the interaction capacity of peptoids to target cysteine proteases [57]. An interesting CZ inhibitor (66, pKi = 6.8) was identified, although not as potent as the analog synthesized by [1][54] (pKi = 7.2). Furthermore, docking experiments suggested that, except for benzyl, the S1 pocket of CZ might accept the bulk of the investigated groups [55]. In S3, there is a fit for biphenyl groups with an interesting proximity between the second ring of two hydrophilic residues Asn70 and Ser61, characterizing probably hydrogen-π interactions (Figure 22).
In another approach concerning nitrile warheads, Burtoloso et al. explored odanacatib (compound 67), a classical cathepsin K inhibitor, as a lead. CZ inhibitors were successfully synthesized, albeit this activity was not translated into in vitro effectiveness. In addition to the observations on cathepsin K inhibitors based on cyclohexane dicarboxamide and odanacatib, scaffolds being bound to their target similarly, three interesting compounds were obtained, with 68 reaching a pEC50 of 6.9, and 68 and 69 reaching a pKi of 5.6, and 70 a near pKi of 5.5 (Figure 23). Compounds 68, 69 and 70 showed higher levels of antitrypanosomal activity than expected for CZ inhibitors. As Burtoloso et al. studied, potency toward CZ tends to increase with molecular size, despite 68 being an exception to this trend [59]. Compound 68 and its R-enantiomer have a contrasting activity probably due to mirrored orientation of S3 moieties. Although the compounds synthesized derived from a covalent inhibitor and present warheads were proved to be potentially covalent in the past, no study was performed from this perspective, but it was thought to be worth mentioning.

In the search for new compounds, new drug discovery methodologies have been developed and used. Gomes et al., for example, explored some prototypes in which the substitution of biphenyl for pyrimidine in P3 caused a change in selectivity towards hCatL based on the previous works [60]. Also, when biphenyl was substituted for phenyl, 0.4 log units of activity were lost (Figure 23). Furthermore, they investigated how inversion of configuration in P3 and replacement of pyridyl with benzyl were not additive, and further cellular testing did not allow any further conclusion. On the other hand, the substitution of cyclopropyl for benzyl increased the activity. A configuration inversion in P3 of the carbons also caused a change in affinity (6.7 for R and 7.4 for S). Among all the synthesized compounds, 74 presented a pKi of 9.2 and an interesting selectivity when compared to Cathepsin L (Δpki = 3.4). As they observed, P3 accommodated and proved tolerable to phenyl and biphenyl groups and P1 benzyl. The replacement of phenyl by biphenyl at P2 caused an increase in pKi, while the replacement of cyclopropane by benzyl methylene increased pKi for the S configuration, but decreased it for the R enantiomer. Regarding P2, the substitution of Leu for Phe decreased the affinity in some cases, and the substitution in P3 by halogenated compounds proved to be tolerable. Nevertheless, it is important to note how this kind of compound could be translated into therapeutics.
Nitrile-containing compounds proved to be bioavailable [62]. The authors used the compounds synthesized by Beaulieu et al. to obtain in vivo and in vitro activities, observing a great correlation between enzyme potency and antiparasitic activity [61]. The most remarkable results were shown compounds cz007 (75) and cz008 (76) (Figure 24). In another approach using nitriles as warheads, Silva et al. performed a comparison study among different warheads [63]. This study had the objective of evaluating warheads based on a carbon-nitrogen double bond. The reference compound for the study and the compounds obtained by lead optimization are depicted in Figure 24.
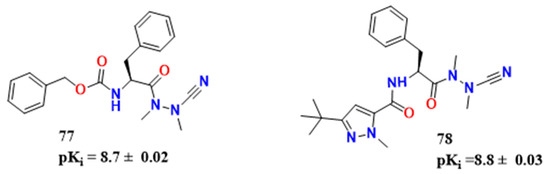
Figure 24. Lead and optimizations obtained by [63].
Silva et al. studied vinyl sulfones, generally irreversible covalent inhibitors, and vinyl amides and esters were synthesized to test whether the replacement of sulfonyl with a less strongly electron-withdrawing group would lead to a reversible inhibition of CZ [63]. The vinyl ester 72 (kinact/Ki = 9.8 × 103 s−1M−1) (Figure 23) was the compound tested for irreversible covalent mechanism, although it was less active than K-777 (79) (kinact/Ki = 5.1 × 105 s−1 M−1). Vinyl esters and amides did not offer any advantage towards more potent nitriles once they were of higher molecular weight and would also be associated with poor pharmacokinetics. In this context, aza-peptide nitriles have been shown to be stable inhibitors of papain-like cysteine proteases and appeared to be useful in activity-based problem regarding T. brucei in the past [31][64] Compound 77 (Figure 24) has a pKi of 8.7 ± 0.02 (Figure 24). This compound is interesting once it lacks the hydrogen bond donor in P1 of typical CZ inhibitors. Compound 77 had the best results in cell-based assays, with a pEC50 of 5.3. However, it also presented the higher pCC50 values 5.0. Azapeptide and aldehyde can be more potent than the traditional dipeptide nitriles.
With the purpose of explaining why some inhibitors are reversible or irreversible, Silva et al. performed quantum mechanics/molecular mechanics studies in addition to a synthesis of the 79 analog, Neq0682 (80) (Figure 25) [65]. QM/MM analysis suggested that His162 plays a significant role as a polarizing residue, making it easier for Cys25 to attack the triple bond, being the energetic barrier very similar for both, 79 and 80 (Figure 25) which is very similar to previously shown in Figure 20. The explanation for the reversibility of the process is that the energetic barrier from the 79-CZ complex is bigger than the 80 one, which was demonstrated by the higher negative value of ΔG in the first reaction.
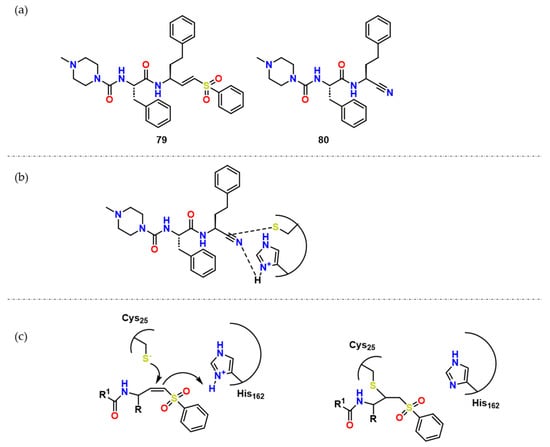
Figure 25. (a) Compound 79 and its analogue 80; (b) Possible network of interactions regarding compound 80; (c) Possible mechanism of action of covalent inhibitors and CZ.
Non-Pepditic Compounds
Non-peptide compounds have been also studied by Jimenez et al. They explored N-acyhydrazones (NAH) and benzenesulfonyl (BS) as groups with potential antitrypanosomal activity totaling 14 compounds designed [66], obtaining compounds with activity, having in common the presence of strong electron-withdrawing groups in para-position on the benzene ring.
Maldonado et al. substituted the sulfonyl by carbonyl group to obtain new N-propionyl N′-benzeneacylhydrazone derivatives (83 and 84) and evaluated their trypanocidal activity (Figure 26) [67]. As a result, compound 83 presented an interesting activity against NINOA strain, and compound 84 presented LC50 comparable to Bzn.

Natural Compounds
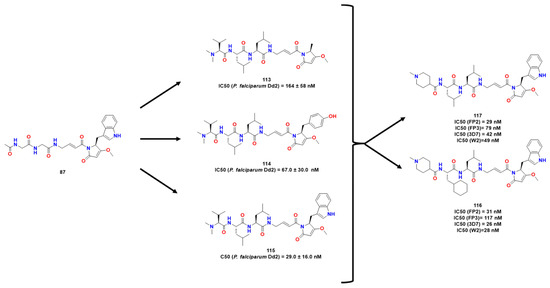
Silva et al. explored naphthoquinone analogs known to exhibit many biological properties, such as anticancer and antitrypanosomal [70]. In this work, they performed a virtual screening on CZ and RD. Thus, 14 molecules were identified as potentially active and were further synthesized. Compound 87 (Figure 27) was a hit compound known as a time-dependent inhibitor, and active in CZ and RD as compounds 88 and 89.

After performing SAR analysis to identify the most promising molecular features of the compounds, they observed through reversibility assay by dilution that only 87 exhibited covalent inhibitory properties, so its mechanism was investigated using covalent molecular docking and molecular dynamics. Therefore, this compound was studied as possible covalent inhibitors of RD. According to the results of the molecular dynamics, 87 established contact with His129 and Cys25. MM-PBSA calculations revealed that van der Waals forces are the most significant to stabilize it. Compound 87 has also been shown to have a strong affinity for RD, which is supported by its low IC50 value. This compound might undergo a nucleophilic attack by Cys25 residue through a C3-Michael addition, as demonstrated by covalent molecular docking and DFT simulations. To assess the reversibility of such molecules, they performed a dilution experiment in which they observed the remaining enzyme inhibition upon dilution for a known irreversible cysteine protease inhibitor E-64, but the same was not observed for 87.
In a different approach, Boudreau et al. chose to investigate analogues and derivatives of gallinamide A (compound 90), an isolated cyanobacteria compound [68]. In this instance, one of the compounds was shown to have a LD50 of 5.1 ± 1.4 nM, while compound 90 had a LD50 of 14.7 nM against Trypanosoma cruzi. Following this, Silva et al. found compounds 92, 93 and 94, whose activities exceeded 90 by 15 times [69]. The structural difference between 92 and 93, in comparison with 94, was the lack of CH2-CH2-Ph in P1, which excludes the stabilizer effect of intramolecular interactions between indole and CH2-CH2-Ph. They also performed calculations on per-residue free energy decomposition-structure ensemble.
Similarly to [68], Silva et al. approached the CZ inhibitory potential of these compounds [69]. As a result, they explored P4 to module pharmacokinetics properties. Simulations of the interaction of 94 with CZ showed that indole in this compound can occur in two principal conformations: interacting with Trp184 and establishing it, and stacked with CH2-CH2-Ph in P1. In the cocrystal, 92 and 94 presented similar activity toward CZ, which might suggest a gain in the binding enthalpy due to intermolecular interactions of the indole ring in 93, comparable to the loss of entropy associated with intermolecular interactions of the indole ring in 94.
2.2.2. Other Targets Related to CD
Proline Racemase [PRAC]
Proline racemase catalyzes the interconversion of L- and D-proline enantiomers [71] with two different residues directly involved in isomerization: Cys130 and Cys270 [72]. This is important for several processes in cell function, including T. cruzi wall construction and immune escape. Also, for its presence in every single stage of T. cruzi cycle and since the parasite is no longer viable after knocking this gene down and gets more virulent if it is overexpressed, this target can be promising [73].
After identifying several isoforms of T. cruzi proline racemases, Berneman et al. chose 2-pyrrolecarboxylic acid (PYC) as a starting point as it is a known PRAC inhibitor with a deficit in water solubility [72]. This latter feature was the focus of the group and led to the synthesis of several pyrazole inhibitors that showed to be more soluble, but that did not present a good affinity for PRAC. In another approach, they performed virtual screening experiments and constructed a QSAR model, leading them to two novel, poorly reactive Michael acceptor compounds: OxoPa (95) and BrOxoPA (96) (Figure 28).

A few years later, Amaral et al. performed crystallographic studies of the two aforementioned compounds [74]. From this data, they proposed new molecular modifications, synthesizing an optimized molecule (NG-P27—97). Unlike 95 and 96, 97 displayed effects on parasite cultures. Both compounds, 95 and 96, exhibited dose-dependent trypanocidal activity for the CL and Y parasite strains. While multiple additions of benznidazole [three times] increased parasite growth inhibition, no cumulative effect was observed after single or multiple treatments with 97 by Amaral et al. [74]. The same group carried out crystallographic studies, and indicated the proximity of the cysteine group towards the double bond in compound 97 (2 Å, approximately) (Figure 28).
Melo et al. expanded the study and synthesized three other derivatives of 97 in order to optimize some pharmacokinetic features for testing against parasites [75]. These compounds had no inhibitory activity against other cysteine proteases. Among these compounds, 98 (Figure 28) was chosen for in vivo assays and had more efficacy in live parasites than the reference compound Bzn.
Trypanothione Reductase (TR)
TR is an essential enzyme for the trypanothione-based thiol metabolism, as stated before for Trypanosoma brucei.
Beig et al. employed in vitro and in silico drug discovery methodologies, and 82 novel inhibitors showed activity in the range of nanomolar against T. cruzi TR (Figure 29) [76]. The best three hits were also relatively active in cell culture (IC50 < 2 μM, comparable to Nfx) and shared an aromatic core ring. Two hits (101 and 102) are shown in Figure 30, and the authors performed experiments that suggested these molecules might act as reversible inhibitors of TR. More details about TR and the structural insights of this protein are described in the reviews written by Tiwari et al. [16] and Battista et al. [77].

Figure 29. Hit compounds described in the reference [76].

2.3. Malaria
P. falciparum has developed resistance to almost all antimalarial drugs used in prevention and treatment. Although most cases are in underdeveloped countries, globalization has made it possible for malaria to spread specially when left uncontrolled [80].
2.3.1. PfGAPDH
GAPDH catalyzes the conversion of glyceraldehyde 3-phosphate (G3P) into 1,3-biphosphoglycerate (1,3-BPG), which is the sixth stage of glycolysis, reducing NAD+ to NAD through a catalytic Cys153. Its potential as a parasitic target comes from the fact that, mainly at the amastigote stage, glycolysis makes up every amount of energy storage for the parasite [78].
PfGADPH is inactivated by compounds capable of alkylating thiols such as 3-bromo-isoxazolines. In this regard, Bruno and coworker decided to analyze the potential activity of avicin and Br-avicin, as well as some of their analogs, and how strong and effective bromine warheads were for PfGADPH inhibition [78]. From their series, two compounds presented the best results: 103 and 104 (Figure 30), with the latter presenting the best covalent parameters (Kinact/Ki = 10.7 ± 2.8). Docking experiments suggested that the isoxazoline moiety, which was near His180 and Tyr323, probably interacted with NAD+ through π-stacking interactions (Figure 31). Furthermore, carbonyl oxygen carried out another hydrogen interaction with a ribose hydroxyl group, while the amine group is over sugar without any other contact. Cys153 was responsible for the nucleophilic attack, later confirmed by UV-vis spectrophotometric detection. It is worth to note that while the glycolytic function of PfGAPDH is inhibited by heme binding this function in enzyme from mammalian is not, which is indicative of difference in the catalytic center [79][81].

Figure 31. Molecule 103 in the active site post-docking carried out by [78]. There are π-stacking interactions between Tyr123 and His1na with molecule 103, which facilitates the nucleophilic attack of Cys153 on carbon 3, with the subsequent leave of bromine. 103 possible interactions with PfGAPDH, as shown in docking studies [4].
In an effort to further investigate this area, Cullia and colleagues expanded on the previous SAR study [4]. They improved upon earlier compounds by modifying the amino acid component, inspired by a group of L-glutamine-dependent amidotransferase inhibitors [78]. Time- and concentration-dependent experiments were conducted with PfGADPH inhibitors, and compound 103 (Figure 30 and Figure 31) showed the most promising results for inhibiting the protein. The authors attributed this to the presence of a minor substituent linked to the ester group, which improved the compound orientation towards Cys153 from either the Pi or Ps site. In addition, the amino group interacted with Thr154 and Gly215 through hydrogen bonds, while the methyl ester group imitated the location of the phosphate substrate in P1 (Figure 31). This orientation facilitated bromine release and subsequent enzyme inactivation. However, this trend of activity was not observed in antiparasitic activity, which the authors suggested was due to the essential role of the α-carbonyl group in transporter recognition. The authors also highlighted compounds 105 and 106, which showed lower activity than 103 but exhibited considerably lower toxicity, with selectivity indexes greater than 175 and 357, respectively.
Galbiati and colleagues aimed to improve the metabolic stability and toxicity of earlier compounds containing ester and amide moieties, which hydrolyze rapidly into carboxylic acid in biological media [82]. They substituted the ester and amide groups with bioisosteres such as oxadiazole, triazole, oxazole, thiazole, and thiadiazole and evaluated GAPDH inhibition. Compound 107 was found to be the most promising of the series, Achieving submicromolar activity for both strains of P. falciparum. These new compounds also had higher lipophilicity, which may contribute to increased membrane penetration compared to the initial compounds, with logD values of 1.41 for compound 105, 0.76 for compound 106, and 3.35 for compound 107. The authors stressed the synthetic simplicity of these novel compounds and suggested that further studies, including with PfGAPDH itself, could be conducted. For information on other types of compounds and inhibitors related to 3-halo-4,5-dihydroisoxazole, refer to [83].
2.3.2. Falcipains
There are four papain-like cysteine proteases characterized for P. falciparum named falcipains, FP-1, FP-2 and FP-2′, FP-3. FP-2 and FP-3 diverge by 37% in amino acid sequence and are expressed in the acidic vacuoles of schizonts and trophozoites. Studies have shown that inhibiting these two latter enzymes prevents hemoglobin hydrolysis and parasite development. Also, knocked-out experiments concerning FP-2 led to the intermittent hindrance of hemoglobin hydrolysis in trophozoites, which made these parasites susceptible to inhibitors, although metabolic recovery was observed due to the probable expression of FP-3 [84].
In light of the studies of these targets, Ettari et al. have conducted a research on the construction of new peptidomimetics with a 1,4-benzodiazepine scaffold introduced into a dipeptide moiety, simulating a sequence of peptides necessary to bind mainly with FP-2 (Figure 32) [84].

Figure 32. Studies performed by Ettari et al. [85].
From previous studies, Ettari et al. studied the effects of modifications in some parts of the structures present in 108 [86]. The methyl group at P1′ was replaced, which maintained irreversible inhibition but significantly reduced affinity and inhibitory potency. This difference was attributed to the divergence in lipophilicity of the compounds. Docking experiments provided further explanation, and the k2 values displayed by the compounds indicated a disfavoring of covalent adduct formation. While the carbonyl group of these compounds seemed to interact with the Trp206 side chain through hydrogen bonding, the P1′ substituent showed a different orientation. In compound 108, the methyl group was directed towards Trp206, whereas in compound 109, the groups were in proximity to residues Asn173, Val152, and Trp206, respectively, and the small distance between double bond and the sulfur atom indicating possible Michael addition. The removal of chlorine from the distal aryl moiety resulted in a derivative with reversible activity and low affinity for falcipain. Molecular dynamics studies suggested that the presence of chlorine may have contributed to a more stable conformation of compound 108 [86].
Previti et al. continued these lines of research by synthesizing a new series of peptide-based compounds as potential FP-2 and RD inhibitors, using compound 21 and k11002 (110) as lead compounds (Figure 33) [87]. The interaction of the compounds with FP-2 was lower than that presented for RD as cited previously here. They then tested several warheads and observed a trend of reactivity that followed this order: ketones > sulfones > esters > nitriles, with the vinyl ketones presenting the most potent inhibitors studied by Previti et al. (111 and 112; IC50 of 0.25 and 0.11 μM/EC50 of 3.16 ± 1.14 and 2.74 ± 1.02 μM P. falciparum respectively) (Figure 34).
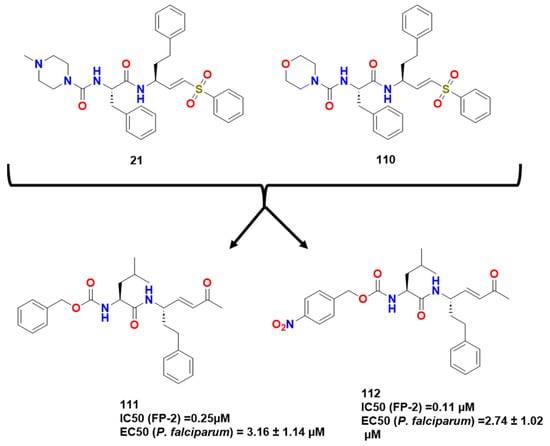
Figure 33. Hit compounds synthesized by Previti et al. [87].
Natural Compounds
With the purpose of taking advantage of natural compounds bearing privileged structures, Conroy et al. reported the synthesis and biological assay of gallinamide A [89] after its previous isolation [89]. From these efforts, the authors demonstrated interesting in vitro activity against P. falciparum with an IC50 of 50 nM of compound 90 and no harmful effect on red blood cells. They observed that replacing N-acyl pyrrolidine group had a negative impact on activity against FPs and parasites and assessed whether one or both olefinic moieties were necessary for the inhibition. They observed that the reduction in acyl pyrrolidine or in the α,β-unsaturated portion caused a detrimental effect in antiplasmodial activity.
Also, the substitution of N-acyl pyrrolidine with other C-terminal groups results in a loss of activity. The derivatives in Figure 35 presented interesting activity against P. falciparum. Substitution of N-acyl pyrrolidine was attempted with benzylamide, although it was not successful. They attributed the importance of this moiety to a Michael acceptor for the active-site Cys residues in FPs in comparison with the corresponding amide. Chain extension was also attempted but failed due to the loss of activity [89].

Considering both compounds 107 and 108, Stoye et al. (Figure 34) proposed bioisosteric replacements of the ester bond for an amide bond in an attempt to stabilize the compounds metabolically [88]. Moreover, the authors maintained indole-derived pyrrolidone present in the gallinamide A, which possessed significant activity towards RD. Then, a target library was constructed, and the compounds were analyzed against FP1 and FP2 and R1 variation appeared to be well-tolerated. The half-lives of plasma and blood were increased by sterically hindered substituents in 1 and 3. Thus, they assessed how 117 acted against some strains of Plasmodium falciparum, being more potent than its previous leads and was evaluated against mice infected with Plasmodium. Further studies might be done for deeper conclusions.
2.3.3. Proteasomes
Proteasomes are responsible for regulating specific processes within the cell cycle. Its composition consists of 20S catalytic units, which embrace 2 heptameric rings of beta subunits. The β1 unit behaves as caspase-like structures (ceasing itself after acidic residues), while the β2 subunit presents trypsin-like activity (cleaves after basic residues) and β5 subunit, chymotrypsin-like activity [92].
Proteasome inhibitors have potential activity against Malaria. Also, they have been discovered to be the cause of apoptosis in certain tumor-derived cells, which made their appearance in investigations against some types of cancer [92]. Furthermore, they can be active against artemisinin-sensitive and resistant parasites.
Benzoxaboroles
In past endeavors, epoxyketone and vinyl sulfone warhead-based inhibitors were synthesized and bound irreversibly to proteasome active sites. Bortezomib and ixazomib are two reversible boronate-based inhibitors used clinically to treat multiple myeloma. Bortezomib presents activity against P. falciparum, albeit its activity is not entirely linked to proteasome inhibition. In addition, boronic acid derivatives are known to be serine proteases covalent inhibitors [93] and, usually, boronate peptides are considered for their properties as covalent inhibitors as shown by Groll et al. [92]. For this reason, Xie et al. used direct evolution and screening from a boronate peptide library of human proteasome inhibitors from Takeda Oncology Company to identify inhibitors of the growth of P. falciparum in vitro [90].
Four compounds had good antiplasmodial activity (Figure 36), but compound 121 presented the best selectivity against β2 site comparing plasmodium and human structures (Ki (Pfβ2) = 6.4 nM/Ki (Hβ2) = 487 nM). Compound 120 presented similar t½ at the human β5c site (110 min) concerning dissociation from β5 site (106 min, Ki = 0.1 nM], but only 2 min (Ki = 490 nM) at the B2c site, indicating its preference for β5c site. As opposed to it, 120 had an interesting affinity against β2 and β5 sites of Pf20S (~5 nM) and longer t1/2 (~65 min). After crystallographic assays with pf20s and bortezomib, a covalent bond between threonine and boronate was observed (Figure 36). The boronic acid derivative moiety gave hard-atom oxygen a good spot to react with as Lewis hard-soft theory states. In addition, the amine present in the threonine performed hydrogen interactions with hydroxyl from boronate, and the Gly47 also performed this interaction (Figure 36).

Following these studies, Xie and collaborators attempted to enhance the oral bioavailability of MPI-1 (compound 118) and synthesized a series of compounds with a single amide-bond backbone [90][91]. As a result, they obtained 122 (Figure 36), an orally bioavailable antimalarial proteasome inhibitor with in vivo efficacy. As drawbacks and points of improvement, it was observed enhancement of in vivo half-life and possible inhibition of human T cell activation. Nevertheless, this series demonstrated the potential for a proteasome inhibitor to be used for Malaria in treatment, transmission-blocking, and chemoprophylaxis scenarios, especially when resistance to artemisinin limits the efficacy of existing combinations.
In a cryo-electron microscopy (EM) experiment, the same authors observed interesting contacts between 121 and the active site of Pf20S (Figure 35). The continuous density observed between Thr1 and boronate derived structures was the main feature observed. Another point to highlight was the great number of hydrophobic interactions, particularly near the biphenyl ring.
As it was observed in these assays, the size of the P1 substituent was directly related to potency and selectivity. When phenyl was replaced by biphenyl, 121 was a 5 nM inhibitor of Pfβ5 and a 21-nM inhibitor of 3d7 culture growth. As it was observed, this series presented favorable physico-chemical properties. For further studies, 121 and 122 were assessed for in vivo efficacy. Also, cryo-EM was performed in combination with MPI-5 and bortezomib. From this structure, it could be inferred that MPI-5 performed mainly hydrophobic interactions, especially considering biphenyl moiety, which probably accounted for the gain in potency in comparison with derivatives that only present monophenyl structure.
Also, from further characterization, it was observed that the covalent bonding to bortezomib is 17 times faster that the one characterized by 121 for HsB5c, while the latter residence time is nearly half of the bortezomib in Hs20s. As opposed to this, the rate of dative bond formation on Pf20S is higher than for bortezomib and similar for bortezomib and Hs20 beta 5C, which is consistent with a 14-fold higher inhibition of bortezomib with Hs20s beta 5c and 2.2-fold less potent pf20s beta5 in comparison to 121. In conclusion, they could infer that 122 is a good example of orally bioavailable antimalarial compound presenting in vivo efficacy. However, it would be interesting to improve its half-life in vivo further.
In a separate study, Li et al. used liquid chromatography tandem mass spectrometry to screen 228 synthetic tetradecapeptides [94]. This resulted in 124, in which they discovered a density continuity between this compound and two of P. falciparum 20S, as well as significant selectivity over the human enzyme, and 123 (Figure 37). These compounds explore bulky SAR in many positions in β2 and β5 subunit active sites of the proteasome.

As a continuation of this study, Stokers et al. explored whether 123 and 124 were interesting compounds against P. falciparum resistant strains (Figure 38) [95]. They observed that both had nanomolar potency against diverse parasites. Also, they would state that both of them seem to be more active against the schizont form of the parasite and early ring stages, which is interesting as most of the antimalarial compounds had been focused on the trophozoite stage.

Fk506-Binding Protein 35 (PfFKBP35)
The recruitment of PfFKBP35 is essential for immunosuppression carried out by inhibitors of calcineurin and mTOR [96]. Bianchin et al. offered insights through crystallographic studies of the interactions of PfFKBP-rapamycin [85]. They mentioned the differences between Plasmodium and human FKBP, highlighting the presence of Cys106 and Ser109 instead of His87 and Ile90 in humans (Figure 39). As a result, they confirmed that Cys106 might be investigated as a particular target for covalent inhibition, which was then done through QM/MM research by [97].
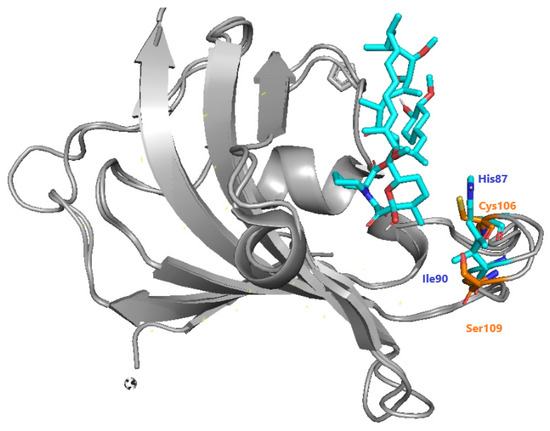
Figure 39. Specific differences between hFKBP (blue) and PfFKBP (orange), adapted from Bianchin et al. [85].
In studies carried out by Atack et al., similar antiplasmodial activity was observed between FK506 (compound 125) and rapamycin (compound 126) [98]. Two interesting compounds were obtained, 127 and 128, presenting activity similar to that of rapamycin (1.4 and 1.9 vs 1.1 μM of IC50, respectively) in addition to the low cytotoxicity in HEK293T cell assays (Figure 40).

Figure 40. Molecules from Atack et al. [98].
This entry is adapted from the peer-reviewed paper 10.3390/ph16071028
References
- Neglected Tropical Diseases. Available online: https://www.who.int/news-room/questions-and-answers/item/neglected-tropical-diseases (accessed on 18 March 2023).
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Kulkarni, S.; Urbahns, K.; Spangenberg, T. Targeted Covalent Inhibitors for the Treatment of Malaria? ACS Infect. Dis. 2020, 6, 2815–2817.
- Cullia, G.; Bruno, S.; Parapini, S.; Margiotta, M.; Tamborini, L.; Pinto, A.; Galbiati, A.; Mozzarelli, A.; Persico, M.; Paladino, A.; et al. Covalent Inhibitors of Plasmodium falciparum Glyceraldehyde 3-Phosphate Dehydrogenase with Antimalarial Activity in Vitro. ACS Med. Chem. Lett. 2019, 10, 590–595.
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017, 22, 3–20.
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human African trypanosomiasis: Pharmacological re-engagement with a neglected disease: Drugs for human African trypanosomiasis. Br. J. Pharmacol. 2007, 152, 1155–1171.
- Sorci, G.; Faivre, B. Inflammation and oxidative stress in vertebrate host–parasite systems. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 71–83.
- Leroux, A.E.; Krauth-Siegel, R.L. Thiol redox biology of trypanosomatids and potential targets for chemotherapy. Mol. Biochem. Parasitol. 2016, 206, 67–74.
- Benítez, D.; Franco, J.; Sardi, F.; Leyva, A.; Durán, R.; Choi, G.; Yang, G.; Kim, T.; Kim, N.; Heo, J.; et al. Drug-like molecules with anti-trypanothione synthetase activity identified by high throughput screening. J. Enzyme Inhib. Med. Chem. 2022, 37, 912–929.
- Fueller, F.; Jehle, B.; Putzker, K.; Lewis, J.D.; Krauth-Siegel, R.L. High Throughput Screening against the Peroxidase Cascade of African Trypanosomes Identifies Antiparasitic Compounds That Inactivate Tryparedoxin. J. Biol. Chem. 2012, 287, 8792–8802.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42.
- Tovar, J.; Cunningham, M.L.; Smith, A.C.; Croft, S.L.; Fairlamb, A.H. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: Effect on parasite intracellular survival. Proc. Natl. Acad. Sci. USA 1998, 95, 5311–5316.
- Lu, J.; Vodnala, S.K.; Gustavsson, A.-L.; Gustafsson, T.N.; Sjöberg, B.; Johansson, H.A.; Kumar, S.; Tjernberg, A.; Engman, L.; Rottenberg, M.E.; et al. Ebsulfur Is a Benzisothiazolone Cytocidal Inhibitor Targeting the Trypanothione Reductase of Trypanosoma brucei. J. Biol. Chem. 2013, 288, 27456–27468.
- Koch, O.; Cappel, D.; Nocker, M.; Jäger, T.; Flohé, L.; Sotriffer, C.A.; Selzer, P.M. Molecular Dynamics Reveal Binding Mode of Glutathionylspermidine by Trypanothione Synthetase. PLoS ONE 2013, 8, e56788.
- Marcu, A.; Schurigt, U.; Müller, K.; Moll, H.; Krauth-Siegel, R.L.; Prinz, H. Inhibitory effect of phenothiazine- and phenoxazine-derived chloroacetamides on Leishmania major growth and Trypanosoma brucei trypanothione reductase. Eur. J. Med. Chem. 2016, 108, 436–443.
- Tiwari, N.; Tanwar, N.; Munde, M. Molecular insights into trypanothione reductase-inhibitor interaction: A structure-based review. Arch. Pharm. 2018, 351, 1700373.
- Wagner, A.; Le, T.A.; Brennich, M.; Klein, P.; Bader, N.; Diehl, E.; Paszek, D.; Weickhmann, A.K.; Dirdjaja, N.; Krauth-Siegel, R.L.; et al. Inhibitor-Induced Dimerization of an Essential Oxidoreductase from African Trypanosomes. Angew. Chem. Int. Ed. 2019, 58, 3640–3644.
- Kerr, I.D.; Wu, P.; Marion-Tsukamaki, R.; Mackey, Z.B.; Brinen, L.S. Crystal Structures of TbCatB and Rhodesain, Potential Chemotherapeutic Targets and Major Cysteine Proteases of Trypanosoma brucei. Tschudi, C.; editor. PLoS Negl. Trop. Dis. 2010, 4, e701.
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl Sulfones as Antiparasitic Agents and a Structural Basis for Drug Design. J. Biol. Chem. 2009, 284, 25697–25703.
- Schechter, I.; Berger, A. On the active site of proteases. III. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem. Biophys. Res. Commun. 1968, 32, 898–902.
- Chenna, B.C.; Li, L.; Mellott, D.M.; Zhai, X.; Siqueira-Neto, J.L.; Calvet Alvarez, C.; Bernatchez, J.A.; Desormeaux, E.; Alvarez Hernandez, E.; Gomez, J.; et al. Peptidomimetic Vinyl Heterocyclic Inhibitors of Cruzain Effect Antitrypanosomal Activity. J. Med. Chem. 2020, 63, 3298–3316.
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Schirmeister, T.; Diederich, F. Optimization of Triazine Nitriles as Rhodesain Inhibitors: Structure-Activity Relationships, Bioisosteric Imidazopyridine Nitriles, and X-ray Crystal Structure Analysis with Human Cathepsin L. ChemMedChem 2013, 8, 967–975.
- Giroud, M.; Kuhn, B.; Saint-Auret, S.; Kuratli, C.; Martin, R.E.; Schuler, F.; Diederich, F.; Kaiser, M.; Brun, R.; Schirmeister, T.; et al. 2 H-1,2,3-Triazole-Based Dipeptidyl Nitriles: Potent, Selective, and Trypanocidal Rhodesain Inhibitors by Structure-Based Design. J. Med. Chem. 2018, 61, 3370–3388.
- Zhang, H.; Collins, J.; Nyamwihura, R.; Ware, S.; Kaiser, M.; Ogungbe, I.V. Discovery of a quinoline-based phenyl sulfone derivative as an antitrypanosomal agent. Bioorg. Med. Chem. Lett. 2018, 28, 1647–1651.
- Ettari, R.; Previti, S.; Maiorana, S.; Amendola, G.; Wagner, A.; Cosconati, S.; Schirmeister, T.; Hellmich, U.A.; Zappalà, M. Optimization Strategy of Novel Peptide-Based Michael Acceptors for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2019, 62, 10617–10629.
- Maiorana, S.; Ettari, R.; Previti, S.; Amendola, G.; Wagner, A.; Cosconati, S.; Hellmich, U.A.; Schirmeister, T.; Zappalà, M. Peptidyl Vinyl Ketone Irreversible Inhibitors of Rhodesain: Modifications of the P2 Fragment. ChemMedChem 2020, 15, 1552–1561.
- Previti, S.; Ettari, R.; Calcaterra, E.; Di Chio, C.; Ravichandran, R.; Zimmer, C.; Hammerschmidt, S.; Wagner, A.; Bogacz, M.; Cosconati, S.; et al. Development of Urea-Bond-Containing Michael Acceptors as Antitrypanosomal Agents Targeting Rhodesain. ACS Med. Chem. Lett. 2022, 13, 1083–1090.
- Previti, S.; Ettari, R.; Di Chio, C.; Ravichandran, R.; Bogacz, M.; Hellmich, U.A.; Schirmeister, T.; Cosconati, S.; Zappalà, M. Development of Reduced Peptide Bond Pseudopeptide Michael Acceptors for the Treatment of Human African Trypanosomiasis. Molecules 2022, 27, 3765.
- Di Chio, C.; Previti, S.; Amendola, G.; Cosconati, S.; Schirmeister, T.; Zappalà, M.; Ettari, R. Development of Novel Benzodiazepine-Based Peptidomimetics as Inhibitors of Rhodesain from Trypanosoma brucei rhodesiense. ChemMedChem 2020, 15, 995–1001.
- Di Chio, C.; Previti, S.; Amendola, G.; Ravichandran, R.; Wagner, A.; Cosconati, S.; Hellmich, U.A.; Schirmeister, T.; Zappalà, M.; Ettari, R. Development of novel dipeptide nitriles as inhibitors of rhodesain of Trypanosoma brucei rhodesiense. Eur. J. Med. Chem. 2022, 236, 114328.
- Yang, P.Y.; Wang, M.; Li, L.; Wu, H.; He, C.Y.; Yao, S.Q. Design, Synthesis and Biological Evaluation of Potent Azadipeptide Nitrile Inhibitors and Activity-Based Probes as Promising Anti-Trypanosoma brucei Agents. Chem. Eur. J. 2012, 18, 6528–6541.
- Ettari, R.; Tamborini, L.; Angelo, I.C.; Grasso, S.; Schirmeister, T.; Lo Presti, L.; De Micheli, C.; Pinto, A.; Conti, P. Development of Rhodesain Inhibitors with a 3-Bromoisoxazoline Warhead. ChemMedChem 2013, 8, 2070–2076.
- Royo, S.; Rodríguez, S.; Schirmeister, T.; Kesselring, J.; Kaiser, M.; González, F.V. Dipeptidyl Enoates As Potent Rhodesain Inhibitors That Display a Dual Mode of Action. ChemMedChem 2015, 10, 1484–1487.
- Royo, S.; Schirmeister, T.; Kaiser, M.; Jung, S.; Rodríguez, S.; Bautista, J.M.; González, F.V. Antiprotozoal and cysteine proteases inhibitory activity of dipeptidyl enoates. Bioorg. Med. Chem. 2018, 26, 4624–4634.
- Ettari, R.; Previti, S.; Cosconati, S.; Kesselring, J.; Schirmeister, T.; Grasso, S.; Zappalà, M. Synthesis and biological evaluation of novel peptidomimetics as rhodesain inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 1184–1191.
- Ettari, R.; Previti, S.; Cosconati, S.; Maiorana, S.; Schirmeister, T.; Grasso, S.; Zappalà, M. Development of novel 1,4-benzodiazepine-based Michael acceptors as antitrypanosomal agents. Bioorg. Med. Chem. Lett. 2016, 26, 3453–3456.
- Steverding, D. On the Reversible and Irreversible Inhibition of Rhodesain by Curcumin. Molecules 2019, 25, 143.
- Schirmeister, T.; Schmitz, J.; Jung, S.; Schmenger, T.; Krauth-Siegel, R.L.; Gütschow, M. Evaluation of dipeptide nitriles as inhibitors of rhodesain, a major cysteine protease of Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2017, 27, 45–50.
- Jung, S.; Fuchs, N.; Johe, P.; Wagner, A.; Diehl, E.; Yuliani, T.; Zimmer, C.; Barthels, F.; Zimmermann, R.A.; Klein, P.; et al. Fluorovinylsulfones and -Sulfonates as Potent Covalent Reversible Inhibitors of the Trypanosomal Cysteine Protease Rhodesain: Structure–Activity Relationship, Inhibition Mechanism, Metabolism, and In Vivo Studies. J. Med. Chem. 2021, 64, 12322–12358.
- McShan, D.; Kathman, S.; Lowe, B.; Xu, Z.; Zhan, J.; Statsyuk, A.; Ogungbe, I.V. Identification of non-peptidic cysteine reactive fragments as inhibitors of cysteine protease rhodesain. Bioorg. Med. Chem. Lett. 2015, 25, 4509–4512.
- Ehmke, V.; Quinsaat, J.E.Q.; Rivera-Fuentes, P.; Heindl, C.; Freymond, C.; Rottmann, M.; Brun, R.; Schirmeister, T.; Diederich, F. Tuning and predicting biological affinity: Aryl nitriles as cysteine protease inhibitors. Org. Biomol. Chem. 2012, 10, 5764.
- Giroud, M.; Dietzel, U.; Anselm, L.; Banner, D.; Kuglstatter, A.; Benz, J.; Blanc, J.-B.; Gaufreteau, D.; Liu, H.; Lin, X.; et al. Repurposing a Library of Human Cathepsin L Ligands: Identification of Macrocyclic Lactams as Potent Rhodesain and Trypanosoma brucei Inhibitors. J. Med. Chem. 2018, 61, 3350–3369.
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice: Essentiality of pteridine reductase 1. Mol. Microbiol. 2010, 77, 658–671.
- Khalaf, A.I.; Huggan, J.K.; Suckling, C.J.; Gibson, C.L.; Stewart, K.; Giordani, F.; Barrett, M.P.; Wong, P.E.; Barrack, K.L.; Hunter, W.N. Structure-Based Design and Synthesis of Antiparasitic Pyrrolopyrimidines Targeting Pteridine Reductase 1. J. Med. Chem. 2014, 57, 6479–6494.
- Saldivia, M.; Fang, E.; Ma, X.; Myburgh, E.; Carnielli, J.B.T.; Bower-Lepts, C.; Brown, E.; Ritchie, R.; Lakshminarayana, S.B.; Chen, Y.-L.; et al. Targeting the trypanosome kinetochore with CLK1 protein kinase inhibitors. Nat. Microbiol. 2020, 5, 1207–1216.
- Lenz, M.; Krauth-Siegel, R.L.; Schmidt, T.J. Natural Sesquiterpene Lactones of the 4,15-iso-Atriplicolide Type are Inhibitors of Trypanothione Reductase. Molecules 2019, 24, 3737.
- Belluti, F.; Uliassi, E.; Veronesi, G.; Bergamini, C.; Kaiser, M.; Brun, R.; Viola, A.; Fato, R.; Michels, P.A.M.; Krauth-Siegel, R.L.; et al. Toward the Development of Dual-Targeted Glyceraldehyde-3-phosphate Dehydrogenase/Trypanothione Reductase Inhibitors against Trypanosoma brucei and Trypanosoma cruzi. ChemMedChem 2014, 9, 371–382.
- Nishino, M.; Choy, J.W.; Gushwa, N.N.; Oses-Prieto, J.A.; Koupparis, K.; Burlingame, A.L.; Renslo, A.R.; McKerrow, J.H.; Taunton, J. Hypothemycin, a fungal natural product, identifies therapeutic targets in Trypanosoma brucei. eLife 2013, 2, e00712.
- Calixto, J.B. The role of natural products in modern drug discovery. An. Acad. Bras. Ciênc. 2019, 91 (Suppl. S3), e20190105.
- Oli, S.; Abdelmohsen, U.; Hentschel, U.; Schirmeister, T. Identification of Plakortide E from the Caribbean Sponge Plakortis halichondroides as a Trypanocidal Protease Inhibitor using Bioactivity-Guided Fractionation. Mar. Drugs 2014, 12, 2614–2622.
- Zhang, H.; Collins, J.; Nyamwihura, R.; Crown, O.; Ajayi, O.; Ogungbe, I.V. Vinyl sulfone-based inhibitors of trypanosomal cysteine protease rhodesain with improved antitrypanosomal activities. Bioorg. Med. Chem. Lett. 2020, 30, 127217.
- Doyle, P.S.; Zhou, Y.M.; Hsieh, I.; Greenbaum, D.C.; McKerrow, J.H.; Engel, J.C. The Trypanosoma cruzi Protease Cruzain Mediates Immune Evasion. PLoS Pathog. 2011, 7, e1002139.
- Lameiro, R.D.F.; Shamim, A.; Rosini, F.; Cendron, R.; Jatai Batista, P.H.; Montanari, C.A. Synthesis, biochemical evaluation and molecular modeling studies of nonpeptidic nitrile-based fluorinated compounds. Future Med. Chem. 2021, 13, 25–43.
- Löser, R.; Schilling, K.; Dimmig, E.; Gütschow, M. Interaction of Papain-like Cysteine Proteases with Dipeptide-Derived Nitriles. J. Med. Chem. 2005, 48, 7688–7707.
- Avelar, L.A.A.; Camilo, C.D.; de Albuquerque, S.; Fernandes, W.B.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; McKerrow, J.H.; Montanari, C.A.; Orozco, E.V.M.; et al. Molecular Design, Synthesis and Trypanocidal Activity of Dipeptidyl Nitriles as Cruzain Inhibitors. PLoS Negl. Trop. Dis. 2015, 9, e0003916.
- Cianni, L.; Sartori, G.; Rosini, F.; De Vita, D.; Pires, G.; Lopes, B.R.; Leitão, A.; Burtoloso, A.C.B.; Montanari, C.A. Leveraging the cruzain S3 subsite to increase affinity for reversible covalent inhibitors. Bioorg. Chem. 2018, 79, 285–292.
- Alves, L.; Santos, D.A.; Cendron, R.; Rocho, F.R.; Matos, T.K.B.; Leitão, A.; Montanari, C.A. Nitrile-based peptoids as cysteine protease inhibitors. Bioorg. Med. Chem. 2021, 41, 116211.
- Dos Santos, A.M.; Cianni, L.; De Vita, D.; Rosini, F.; Leitão, A.; Laughton, C.A.; Lameira, J.; Montanari, C.A. Experimental study and computational modelling of cruzain cysteine protease inhibition by dipeptidyl nitriles. Phys. Chem. Chem. Phys. 2018, 20, 24317–24328.
- Burtoloso, A.C.B.; de Albuquerque, S.; Furber, M.; Gomes, J.C.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; Montanari, C.A.; Quilles, J.C.; Ribeiro, J.F.R.; et al. Anti-trypanosomal activity of non-peptidic nitrile-based cysteine protease inhibitors. PLoS Negl. Trop. Dis. 2017, 11, e0005343.
- Gomes, J.C.; Cianni, L.; Ribeiro, J.; dos Reis Rocho, F.; da Costa Martins Silva, S.; Batista, P.H.J.; Moraes, C.B.; Franco, C.H.; Freitas-Junior, L.H.G.; Kenny, P.W.; et al. Synthesis and structure-activity relationship of nitrile-based cruzain inhibitors incorporating a trifluoroethylamine-based P2 amide replacement. Bioorg. Med. Chem. 2019, 27, 115083.
- Ndao, M.; Beaulieu, C.; Black, W.C.; Isabel, E.; Vasquez-Camargo, F.; Nath-Chowdhury, M.; Massé, F.; Mellon, C.; Methot, N.; Nicoll-Griffith, D.A. Reversible Cysteine Protease Inhibitors Show Promise for a Chagas Disease Cure. Antimicrob. Agents Chemother. 2014, 58, 1167–1178.
- Beaulieu, C.; Isabel, E.; Fortier, A.; Massé, F.; Mellon, C.; Méthot, N.; Ndao, M.; Nicoll-Griffith, D.; Lee, D.; Park, H.; et al. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg. Med. Chem. Lett. 2010, 20, 7444–7449.
- Silva, D.G.; Ribeiro, J.F.R.; De Vita, D.; Cianni, L.; Franco, C.H.; Freitas-Junior, L.H.; Moraes, C.B.; Rocha, J.R.; Burtoloso, A.C.B.; Kenny, P.W.; et al. A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5031–5035.
- Löser, R.; Frizler, M.; Schilling, K.; Gütschow, M. Azadipeptide Nitriles: Highly Potent and Proteolytically Stable Inhibitors of Papain-Like Cysteine Proteases. Angew. Chem. Int. Ed. 2008, 47, 4331–4334.
- Silva, J.R.A.; Cianni, L.; Araujo, D.; Batista, P.H.J.; de Vita, D.; Rosini, F.; Leitão, A.; Lameira, J.; Montanari, C.A. Assessment of the Cruzain Cysteine Protease Reversible and Irreversible Covalent Inhibition Mechanism. J. Chem. Inf. Model. 2020, 60, 1666–1677.
- Elizondo-Jimenez, S.; Moreno-Herrera, A.; Reyes-Olivares, R.; Dorantes-Gonzalez, E.; Nogueda-Torres, B.; Oliveira, E.; Romeiro, N.; Lima, L.; Palos, I.; Rivera, G. Synthesis, Biological Evaluation and Molecular Docking of New Benzenesulfonylhydrazone as Potential anti-Trypanosoma cruzi Agents. Med. Chem. 2017, 13, 149–158.
- Delgado-Maldonado, T.; Nogueda-Torres, B.; Espinoza-Hicks, J.C.; Vázquez-Jiménez, L.K.; Paz-González, A.D.; Juárez-Saldívar, A.; Rivera, G. Synthesis and biological evaluation in vitro and in silico of N-propionyl-N′-benzeneacylhydrazone derivatives as cruzain inhibitors of Trypanosoma cruzi. Mol. Divers. 2022, 26, 39–50.
- Boudreau, P.D.; Miller, B.W.; McCall, L.-I.; Almaliti, J.; Reher, R.; Hirata, K.; Le, T.; Siqueira-Neto, J.L.; Hook, V.; Gerwick, W.H. Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma cruzi Cruzain. J. Med. Chem. 2019, 62, 9026–9044.
- Da Silva, E.B.; Sharma, V.; Hernandez-Alvarez, L.; Tang, A.H.; Stoye, A.; O’Donoghue, A.J.; Gerwick, W.H.; Payne, R.J.; McKerrow, J.H.; Podust, L. Intramolecular Interactions Enhance the Potency of Gallinamide a Analogs against Trypanosoma cruzi. 2021. Available online: http://biorxiv.org/lookup/doi/10.1101/2021.12.22.473926 (accessed on 17 March 2023).
- Silva, L.R.; Guimarães, A.S.; do Nascimento, J.; do Santos Nascimento, I.J.; da Silva, E.B.; McKerrow, J.H.; Cardoso, S.H.; da Silva-Júnior, E.F. Computer-aided design of 1,4-naphthoquinone-based inhibitors targeting cruzain and rhodesain cysteine proteases. Bioorg. Med. Chem. 2021, 41, 116213.
- Buschiazzo, A.; Goytia, M.; Schaeffer, F.; Degrave, W.; Shepard, W.; Grégoire, C.; Chamond, N.; Cosson, A.; Berneman, A.; Coatnoan, N. Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc. Natl. Acad. Sci. USA 2006, 103, 1705–1710.
- Berneman, A.; Montout, L.; Goyard, S.; Chamond, N.; Cosson, A.; d’Archivio, S.; Gouault, N.; Uriac, P.; Blondel, A.; Minoprio, P. Combined Approaches for Drug Design Points the Way to Novel Proline Racemase Inhibitor Candidates to Fight Chagas’ Disease. PLoS ONE 2013, 8, e60955.
- Chamond, N.; Goytia, M.; Coatnoan, N.; Barale, J.-C.; Cosson, A.; Degrave, W.M.; Minoprio, P. Trypanosoma cruzi proline racemases are involved in parasite differentiation and infectivity: Trypanosoma cruzi proline racemases. Mol. Microbiol. 2005, 58, 46–60.
- Amaral, P.d.A.; Autheman, D.; de Melo, G.D.; Gouault, N.; Cupif, J.-F.; Goyard, S.; Dutra, P.; Coatnoan, N.; Cosson, A.; Monet, D. Designed mono- and di-covalent inhibitors trap modeled functional motions for Trypanosoma cruzi proline racemase in crystallography. PLoS Negl. Trop. Dis. 2018, 12, e0006853.
- De Melo, G.D.; Coatnoan, N.; Gouault, N.; Cupif, J.-F.; Renault, J.; Cosson, A.; Uriac, P.; Blondel, A.; Minoprio, P. Prodrugs as new therapies against Chagas disease: In vivo synergy between Trypanosoma cruzi proline racemase inhibitors and benznidazole. J. Glob. Antimicrob. Resist. 2022, 28, 84–89.
- Beig, M.; Oellien, F.; Garoff, L.; Noack, S.; Krauth-Siegel, R.L.; Selzer, P.M. Trypanothione Reductase: A Target Protein for a Combined In Vitro and In Silico Screening Approach. PLoS Negl. Trop. Dis. 2015, 9, e0003773.
- Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924.
- Bruno, S.; Pinto, A.; Paredi, G.; Tamborini, L.; De Micheli, C.; La Pietra, V.; Marinelli, L.; Novellino, E.; Conti, P.; Mozzarelli, A. Discovery of Covalent Inhibitors of Glyceraldehyde-3-phosphate Dehydrogenase, A Target for the Treatment of Malaria. J. Med. Chem. 2014, 57, 7465–7471.
- Bruno, S.; Margiotta, M.; Pinto, A.; Cullia, G.; Conti, P.; De Micheli, C.; Mozzarelli, A. Selectivity of 3-bromo-isoxazoline inhibitors between human and Plasmodium falciparum glyceraldehyde-3-phosphate dehydrogenases. Bioorg. Med. Chem. 2016, 24, 2654–2659.
- Fact Sheet about Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 17 March 2023).
- Hannibal, L.; Collins, D.; Brassard, J.; Chakravarti, R.; Vempati, R.; Dorlet, P.; Santolini, J.; Dawson, J.H.; Stuehr, D.J. Heme Binding Properties of Glyceraldehyde-3-phosphate Dehydrogenase. Biochemistry 2012, 51, 8514–8529.
- Galbiati, A.; Zana, A.; Coser, C.; Tamborini, L.; Basilico, N.; Parapini, S.; Taramelli, D.; Conti, P. Development of Potent 3-Br-isoxazoline-Based Antimalarial and Antileishmanial Compounds. ACS Med. Chem. Lett. 2021, 12, 1726–1732.
- Pinto, A.; Tamborini, L.; Cullia, G.; Conti, P.; De Micheli, C. Inspired by Nature: The 3-Halo-4,5-dihydroisoxazole Moiety as a Novel Molecular Warhead for the Design of Covalent Inhibitors. ChemMedChem 2016, 11, 10–14.
- Mane, U.R.; Gupta, R.C.; Nadkarni, S.S.; Giridhar, R.R.; Naik, P.P.; Yadav, M.R. Falcipain inhibitors as potential therapeutics for resistant strains of malaria: A patent review. Expert Opin. Ther. Pat. 2013, 23, 165–187.
- Bianchin, A.; Allemand, F.; Bell, A.; Chubb, A.J.; Guichou, J.F. Two crystal structures of the FK506-binding domain of Plasmodium falciparum FKBP35 in complex with rapamycin at high resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1319–1327.
- Ettari, R.; Micale, N.; Grazioso, G.; Bova, F.; Schirmeister, T.; Grasso, S.; Zappalà, M. Synthesis and Molecular Modeling Studies of Derivatives of a Highly Potent Peptidomimetic Vinyl Ester as Falcipain-2 Inhibitors. ChemMedChem 2012, 7, 1594–1600.
- Previti, S.; Ettari, R.; Cosconati, S.; Amendola, G.; Chouchene, K.; Wagner, A.; Hellmich, U.A.; Ulrich, K.; Krauth-Siegel, R.L.; Wich, P.R.; et al. Development of Novel Peptide-Based Michael Acceptors Targeting Rhodesain and Falcipain-2 for the Treatment of Neglected Tropical Diseases . J. Med. Chem. 2017, 60, 6911–6923.
- Stoye, A.; Juillard, A.; Tang, A.H.; Legac, J.; Gut, J.; White, K.L.; Charman, S.A.; Rosenthal, P.J.; Grau, G.E.R.; Hunt, N.H.; et al. Falcipain Inhibitors Based on the Natural Product Gallinamide A Are Potent in Vitro and in Vivo Antimalarials. J. Med. Chem. 2019, 62, 5562–5578.
- Conroy, T.; Guo, J.T.; Elias, N.; Cergol, K.M.; Gut, J.; Legac, J.; Khatoon, L.; Liu, Y.; McGowan, S.; Rosenthal, P.J.; et al. Synthesis of Gallinamide A Analogues as Potent Falcipain Inhibitors and Antimalarials. J. Med. Chem. 2014, 57, 10557–10563.
- Xie, S.C.; Gillett, D.L.; Spillman, N.J.; Tsu, C.; Luth, M.R.; Ottilie, S.; Duffy, S.; Gould, A.E.; Hales, P.; Seager, B.A.; et al. Target Validation and Identification of Novel Boronate Inhibitors of the Plasmodium falciparum Proteasome. J. Med. Chem. 2018, 61, 10053–10066.
- Xie, S.C.; Metcalfe, R.D.; Mizutani, H.; Puhalovich, T.; Hanssen, E.; Morton, C.J.; Du, Y.; Dogovski, C.; Huang, S.-C.; Ciavarri, J.; et al. Design of proteasome inhibitors with oral efficacy in vivo against Plasmodium falciparum and selectivity over the human proteasome. Proc. Natl. Acad. Sci. USA 2021, 118, e2107213118.
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456.
- Walker, B.; Lynas, J.F. Strategies for the inhibition of serine proteases. Cell. Mol. Life Sci. 2001, 58, 596–624.
- Li, H.; O’Donoghue, A.J.; van der Linden, W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, C.S.; da Fonseca, P.C.A.; Bogyo, M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236.
- Stokes, B.H.; Yoo, E.; Murithi, J.M.; Luth, M.R.; Afanasyev, P.; da Fonseca, P.C.A.; Winzeler, E.A.; Ng, C.L.; Bogyo, M.; Fidock, D.A. Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLOS Pathog. 2019, 15, e1007722.
- Monaghan, P.; Bell, A. A Plasmodium falciparum FK506-binding protein (FKBP) with peptidyl–prolyl cis–trans isomerase and chaperone activities. Mol. Biochem. Parasitol. 2005, 139, 185–195.
- MacDonald, C.A.; Boyd, R.J. Computational insights into the suicide inhibition of Plasmodium falciparum Fk506-binding protein 35. Bioorg. Med. Chem. Lett. 2015, 25, 3221–3225.
- Atack, T.C.; Raymond, D.D.; Blomquist, C.A.; Pasaje, C.F.; McCarren, P.R.; Moroco, J.; Befekadu, H.B.; Robinson, F.P.; Pal, D.; Esherick, L.Y.; et al. Targeted Covalent Inhibition of Plasmodium FK506 Binding Protein 35. ACS Med. Chem. Lett. 2020, 11, 2131–2138.
This entry is offline, you can click here to edit this entry!