Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mesoporous silica nanoparticles (MSNs) have been advocated as nanocarriers for the treatment of various diseases because of their physicochemical properties and biocompatibility. The use of MSNs combined with therapeutic agents can provide better encapsulation and effective delivery. MSNs as nanocarriers might also be a promising tool to lower the therapeutic dosage levels and thereby to reduce undesired side effects. Furthermore, when combined with imaging compounds for diagnosis, they can be employed as theragnostic agents thus allowing both imaging and therapy using the same nanoparticle.
- drug delivery
- biomedical applications
- sol-gel
- biomedical imaging
1. Introduction
Nanoscience and nanotechnology have developed into the backbone of modern research in last years as a result of the introduction of new and improved technologies. Ranging from basic to applied sciences, with a focus on nanoengineering new materials, [1] nanotechnology encompasses fundamental research, engineering and technology and entails the modeling, construction, characterization and manipulation of matters’ properties at the nano-scale.
Nanomaterials are typically identified as materials with at least one dimension in the range of 1–100 nm [2]. Their physical, chemical and biological properties might be completely different from those of bulk matter [3]. The unique features of these nanostructures are attributed to quantum confinement or surface effects [4], which result in interesting and potentially useful catalytic, magnetic, optical and electrical properties [5]. Consequently, nanomaterials have long been a subject of interest worldwide [6], allowing for the development of a diverse range of advanced materials that brings together the demands of high-tech and scientific applications [3], which are already revolutionizing industries such as electronics, medicine and consumer products [7].
Monitoring the size and shape of nanoparticles can help to improve their performance in specific applications (Figure 1). Similarly, characteristics such as chemical composition, charge and the surface can be altered to suit specific purposes. Additionally, functional groups, hydrophilicity, attachment and conjugation with targeting ligands also comprise the essential parameters that need to be considered [8].
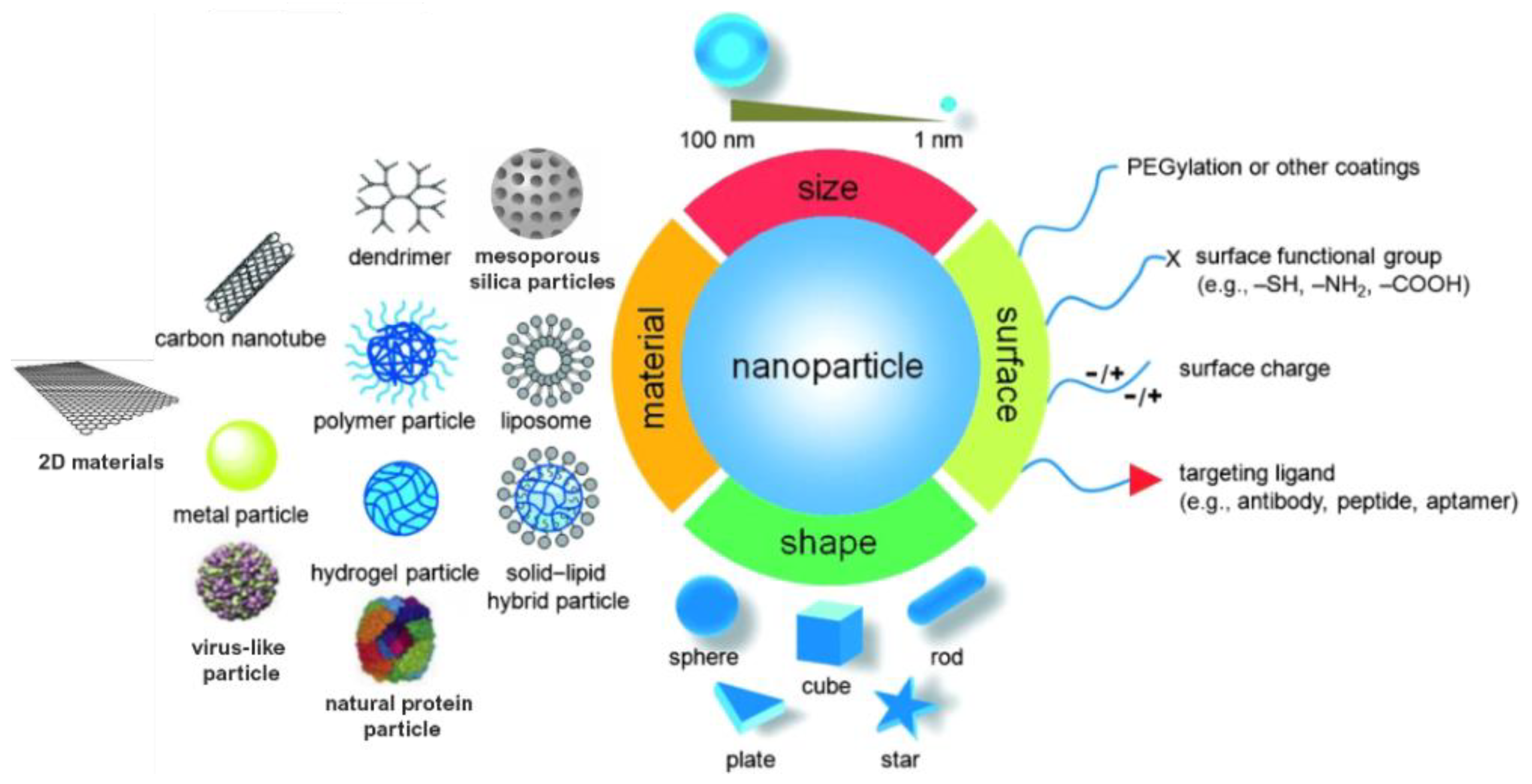
Figure 1. Material, shape, size and surface properties play an important role on the biophysicochemical properties of nanoparticles.
Nanomaterials can be broadly classified according to their organic or inorganic composition. Organic nanomaterials include polymeric nanostructures, dendrimers, lipid bilayers and virus and proteic particles, whereas inorganic nanomaterials are composed of metals, oxides, nitrides, halides, carbonates and chalcogenides, to name some [9]. Among the inorganic materials, nanosized silica has been extensively studied and employed in a wide range of applications [10].
Silicon is widely distributed throughout the world, accounting for ca. 40% of all known minerals. It occurs naturally in a variety of forms in the environment, most frequently in combination with oxygen (both amorphous and crystalline) or hydroxides. Silicon is also found dissolved in the oceans in the form of silicic acid and in living organisms, such as sponges, greases and algae (diatoms) [11]. Silica also comprises one of the most complex and abundant families of materials, occurring naturally in a variety of minerals (including quartz, flint, opal and silicates) and as synthetic products (fused-quartz, fumed-silica, silica-gel, etc.). The Si atom exhibits tetrahedral coordination within the majority of silicates, consisting of four oxygen atoms surrounding a central Si atom [7]. Amorphous silica is primarily prepared through simple manufacturing procedures on a large scale and its properties can be tailored to a wide range of applications. Silica combines a high degree of chemical stability, low toxicity and high biocompatibility with a plethora of possibilities for modifying its physical and chemical properties, as well as the possibility of further modification via functional group coupling, binding of biomolecules or dye doping [12]. Therefore, monodispersed colloidal silica particles with uniform shape, size and composition have demonstrated significant potential, not only in physical chemistry, being used to investigate the dynamic behavior and stability of particulate systems, but also in industries, including pigments, photographic emulsions, ceramics, catalysts, etc. [13].
Porous silica presents a customizable mesoporous structure and pore volume, as well as a high specific surface area. These properties enable the encapsulation of a variety of compounds, including gas and heavy metal ion adsorbents, bioimaging probes and therapeutic agents, such as drugs [14]. In this respect, ordered mesoporous materials, such as the M41S family, comprise an emerging and significant class of inorganic oxides with enormous potential for molecular sieving, catalysis and adsorption processes [5].
Numerous researchers have described various synthetic methodologies for the preparation of mesoporous oxide materials, proposing various mechanisms for the formation of their porous structures. Different factors influence the porous structure formation and affect the size, distribution, and ordering of pores, including precursors (alkoxides, metal salts, etc.), surfactants as structure-directing agents and reaction parameters (pH, temperature, etc.). Their size and shape, which can, for instance, take the form of spheres, rods, or wormlike structures, can also be controlled by adjusting the molar ratio of silica precursors and surfactants, the pH, adding co-solvents or organic swelling agents and introducing alkoxysilane precursors [13][15]. Notably, the fabrication of mesoporous silica nanoparticles is straightforward, scalable, economic and controllable.
2. Silica Nanoparticles
The use of SiO2 nanoparticles (NPs) has increased in recent years, due to the diversification of their properties, thus widening their potential applications. The ease of preparation and surface modification confer the material versatility, being useful in a variety of fields, such as catalysis, semiconductors, pigments, pharmacy, electronics, detergents, cosmetics and sensors. SiO2 NPs can be grown to lead different nanostructures, namely solid nanospheres and mesoporous and hollow silica nanoparticles (HSNs), also adopting diverse morphologies (cubic systems, nanorods, etc.); the characteristics of the resulting particles being closely linked to the synthetic approach.
2.1. Strategies for the Preparation of Silica NPs
Two main approaches, the viz. top-down and bottom-up techniques, have been proposed to obtain silica nanoparticles. In the first one, the dimension of the original size is reduced by breaking down the constituents of bulk materials. Conversely, the bottom-up method is commonly used to produce silica nanoparticles from the atomic or molecular level to the nano-/micro-scale [3]. Due to the advantages of the bottom-up approach, which includes the ability to control the particle size and morphology, this is the strategy most widely employed for the synthesis of inorganic nanostructures, including silica NPs. This protocol also leads to a narrower size distribution through controlling the reaction parameters [1].
2.1.1. The Sol-Gel Method
The sol-gel approach entails the hydrolysis, followed by the condensation of alkoxide monomers into a colloidal solution (sol), acting as the precursor to form an ordered network (gel) of polymer or discrete particles [16][17][18].
In 1956, Kolbe [19] described the formation of silica particles by reacting tetraethyl silicate in an alcoholic solution containing water in the presence of certain bases. It was observed that, using very pure reagents, the reaction, which proceeds slowly, leads to the formation of uniform spherical silica nanoparticles. Later, Stöber et al. [20] developed a system that allowed the controlled growth of spherical silica particles with uniform sizes (50 nm–2 μm) via the hydrolysis of alkyl silicates and their subsequent condensation in low molecular-weight alcohols as the solvent, while using ammonia as the morphological catalyst.
Generally, the formation of silica particles can be divided into two steps, namely nucleation and growth. To describe the growth mechanism of silica, two models have been proposed. The monomer addition model predicts that the particle growth occurs through the addition of other hydrolyzed monomers after an initial nucleation state. The aggregation model describes that primary particles (nuclei) aggregate together to form dimers, trimers and progressively larger particles (secondary particles) [3]. At the same time, the nature of the catalyst strongly influences both the rate and mechanism of hydrolysis and condensation. In the case of an acid-catalyzed reaction, the hydrolysis becomes faster than the condensation, resulting in the formation of numerous small silica particles. Conversely, in the base-catalyzed reaction, the condensation proceeds much faster than the hydrolysis step, leading to larger silica nanoparticles [21]. The acid-catalyzed mechanisms are preceeded by the rapid deprotonation of the substituents bonded to Si, whereas under basic conditions, the hydroxide ion serves as a nucleophile that attacks the silicon atom center of the tetraalkoxysilane. The result of this step is a silanol and an alkoxide ion [9][22].
In particular, the first step of hydrolysis induced by the acid catalyst is the electrophilic attack of the proton on an oxygen atom of the silica precursor, leading to a positive charge on it. This attack makes the bond between the silicon and the attacked oxygen (Si-O) more polarized and facilitates its cleavage, thus producing an alcohol as the leaving group. Therefore, an increase in the water-to-alkoxide ratio increases the rate of hydrolysis [23]. Following the hydrolysis reaction, the condensation reaction occurs immediately [11]. During the condensation, the proton electrophilically attacks the oxygen of the silanol group. This phenomenon causes the oxygen of the silanol to become positively charged and a siloxane bridge is formed because of condensation between both protonated and unprotonated silanol groups [23]. On the other hand, the base-catalyzed hydrolysis proceeds via a nucleophilic attack of the hydroxyl group on the silicon atom of the alkoxide, while the subsequent release of an OR− as the leaving group is facilitated. On condensation, the hydroxyl group of intermediate [Si (OC2H4)4−x (OH)x] reacts with either the ethoxy group of other alkoxysilanes (alcohol condensation) or the hydroxyl group of another hydrolysis intermediate (water condensation) to form Si-O-Si bridges (Scheme 1) [11].
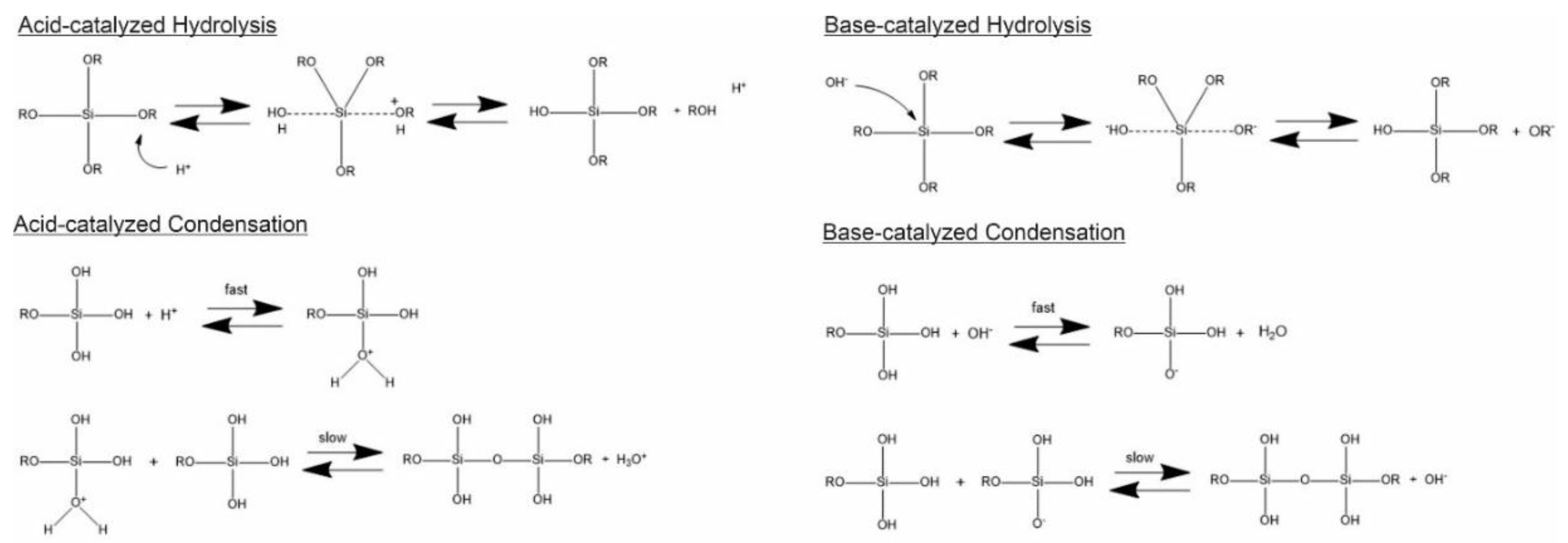
Scheme 1. Hydrolysis and condensation in acidic and basic conditions.
Numerous alkoxysilane precursors (Si(OR)4) have been reported, including tetramethyl orthosilicate (TMOS) or tetraethyl orthosilicate (TEOS) (Figure 2), which undergo hydrolysis and condensation reactions upon specific pH conditions [24].
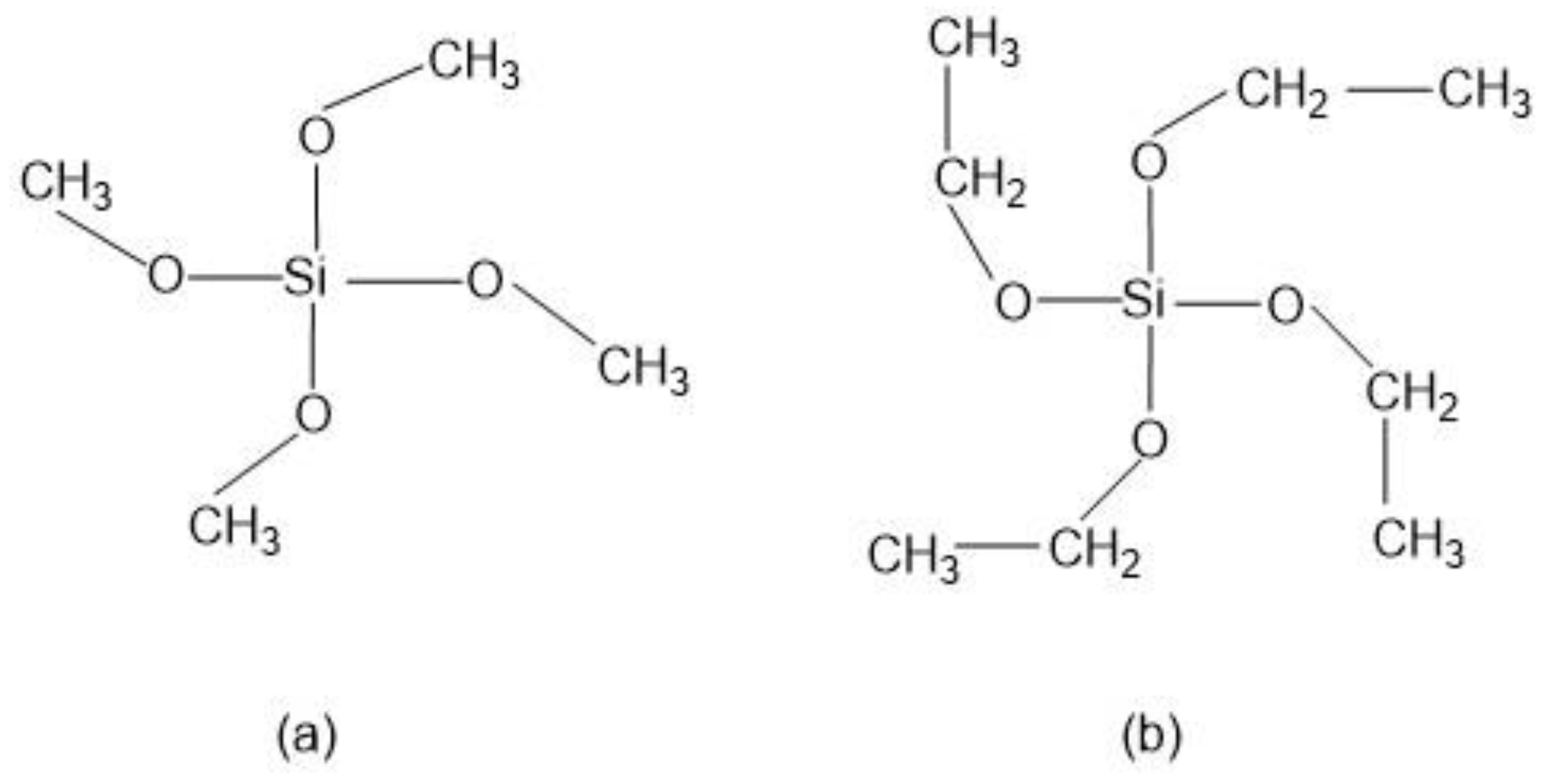
Figure 2. Alkoxysilanes such as (a) tetramethoxysilane (TMOS) and (b) tetraethoxysilane (TEOS) are extensively used to produce silica gels.
The hydrolysis step leads to the generation of silanol groups (Si-OH), the mechanism being mainly conditioned by the catalyst, while its rate depends on the pH, the solvent used and the ratio of water-to-alkoxide. Since alkoxysilanes are not soluble in water, organic co-solvents are required to facilitate hydrolysis. In the condensation step, the silanol group interacts with an alkoxide or silanol group to form a strong siloxane bond (Si-O-Si), thus resulting in the loss of one molecule of alcohol (ROH) or one molecule of water [23].
Silica particles are also synthesized via the sol-gel method using water-in-oil (W/O) emulsions. Reverse micelles in oil reflect the size, shape and size distribution of the particles, as they act as templates for their growth [25]. In the case of reverse microemulsions, the surfactant molecules are dissolved in organic solvents to form spherical micelles. In the presence of water, the polar head groups adopt the lowest energy position and organize to form water-containing microcavities. During the synthesis, silica nanoparticles can be grown inside the reverse micelles by controlling the addition of silicon alkoxides and the catalyst into the medium. The diffusion of the alkoxide into water droplets outcome in hydrolysis of the alkoxide and the formation of alcohol and oxy-hydroxy-silicate species [26]. The main drawbacks of the reverse microemulsion approach are the high costs and the complications associated with the removal of surfactants in the final products [3].
2.1.2. Other Methods
Until now, the sol-gel method has been the most straightforward and effective method for synthesizing monodispersed silica spheres. However, over the years, numerous research groups have introduced improvements in the hydrolysis method to achieve uniform shape and monodisperse distribution [13] and novel routes have been explored for the preparation of silica nanoparticles that have short reaction times, excellent reproducibility and are simple to prepare [27]. Alternative processes worth mentioning include microwave, ultrasound and evaporation-induced approaches, self-assembly (EISA), hydrothermal and solvothermal, or oxidative processes, which have also been explored to optimize the production of pure silica particles [28]. Despite the fact that a detailed explanation of each methodology is outside the scope of the present research, researchers believe that energy-assisted syntheses present some unique benefits.
Energy-assisted approaches, namely the microwave- or ultrasound-induced formation of SiO2 NPs represent attractive, although not generalized, techniques to obtain functional nanostructures. Due to the characteristics of the incident energy source (300 MHz–300 GHz), microwave-assisted synthesis requires almost negligible working times and yields high quality and reproducible NPs (in terms of size distribution and shape). The formation process occurs due to the alignment with the electromagnetic field of the polar and/or charged molecules present within the irradiated system, which, as an effect of the energy, increases its temperature (due to the vigorous agitation of the species). The temperature gradient profile generated during the process is highly homogeneous, favoring aspects as a high reaction yield by employing much shorter times than sol-gel methods or the formation of species with narrow size distributions. The possibility of tuning the characteristics of the incident energy also allows controlling the conditions of the system, thus improving the synthetic approach, such as for instance, reducing the times of the reaction or enhancing the selectivity towards a selected material, therefore minimizing the need of further purification steps for the removal of undesired products. Different Si precursors have been employed to obtain SiO2 nanospheres using microwave, for instance, TMOS in the presence of HCl as a catalyst. Noticeably, the approach requires employing a solvent, such as acetone, which allows the absorption of the incident radiation by the reactants. Thus, the SiO2 NPs were obtained using times as short as 1 min and mild conditions (125 °C). The size of the resulting NPs depends on the concentration, time of treatment and irradiation power [29]. Despite the fact that the protocol can be applied to a wide variety of nanostructures, the main drawbacks are related to the low scalability of the approach or the poor homogeneity of the surface of the obtained NPs [22]. On the other hand, taking advantage of the efficiency of the energy-assisted methodologies, a hybrid approach, consisting on the combination of the well-known sol-gel protocol and ultrasound allowed controlling the size of the core NPs (d = 13 nm), thus confirming the versatility of the technique and the possibility of controlling the morphological characteristics of the products [30].
2.2. Mesoporous Silica Nanoparticles
According to the IUPAC definition, porous materials can be classified into three groups based on their pore sizes, namely microporous, mesoporous and macroporous ones, with corresponding pore diameters of <2 nm, 2–50 nm and >50 nm, respectively [31].
In 1990, Yanagisawa et al. [32] reported the condensation of silicate layers to form a structure of 3D “nanoscale pores” [33]. The protocol, based on the intercalation of alkyltrimethylammonium cations into kanemite silicate, followed by calcination (removal of organic species), led to the development of the characteristic MCM-41 mesoporous materials. On their basis, in 1992, Mobil Corporation Laboratories designated a new family of amorphous and ordered mesoporous inorganic materials with exceptionally large and uniform pore conformations, narrow pore size distributions and large surface areas: the M41S (molecular 41 sieves) [31][33][34]. This family includes two main groups: those with a three-dimensional interconnected porous structure (cubic-ordered pore structure) designated as MCM-48 and those with a one-dimensional and hexagonally ordered pore structure, known as MCM-41 and MCM-50, which have an unstable lamellar structure [17][35]. Following their discovery, significant efforts have been made to control the properties (particularly, the pore size and morphology) of mesoporous silica. Through this research, in 1995, new families of mesoporous silica systems (MSS), namely SBA, MSU and FSM, were developed [36]. Later, in 1998, the search for a mesoporous material with a hexagonal array of pores resulted in the formation of the Santa Barbara Amorphous No.15 (SBA-15), a research gambit in mesoporous material development [18]. SBA-15 demonstrated large pore sizes (up to 30 nm), as well as thermal, mechanical and chemical resistance, establishing it as the preferred mesoporous material for catalysis’ applications [23].
Among the mesoporous silica systems, MSNs offer outstanding advantages over non-porous silica nanoparticles, such as large pore volume (0.6–1.5 cm3/g), high surface area (700–1000 m2/g), tunable particle sizes, tailorable pore diameter (2–10 nm), high thermal, chemical and biological stabilities, ease of functionalization, biodegradability and good biocompatibility [37][38]. MSNs have a high rate of mass transport, a high affinity for substrates and a high dispersibility in solutions [39]. Additionally, they have two functional surfaces, the inner one, which corresponds to the pore channels and the external surface, which is highly susceptible to be selectively functionalized [40]. The particle size, pore diameter and characteristics of the silica NPs depend on the synthesis conditions [41]. Some examples of silica NPs, analyzed by transmission electron microscopy, are illustrated in Figure 3.

Figure 3. Transmission electron microscopy (TEM) images of three spherical MSNs with different particle and pore sizes: (a) 350 nm and 2–3 nm of pore diameter, (b) 150 nm size and 2–3 nm pore diameter and (c) 100 nm and 6–8 nm pore diameters.
Silica spheres possessing nanopores of well-defined size and connectivity are of significant interest, for instance, for the use in catalysis, chromatography, for the controlled release of drugs and as hosts for optically active compounds [42].
2.2.1. Synthesis of Mesoporous Silica NPs
Since their development, a variety of mechanisms have been proposed to explain the formation of MSNs [1]. At the most fundamental level, all of these models imply that supramolecular assemblies of surfactants serve as templates for the formation of porous structures in the inorganic matrix [17].
Grun et al. reported the synthesis of MSNs with multiple dimensions, pore sizes, structures and morphologies using a modified Stöber’s method [20][23]. Constant research has resulted in variations of the synthesis conditions and methods to yield stable and monodisperse MSNs [16]. Based on the previously detailed methodologies, mesoporous silica spheres can be synthetized by introducing templating surfactants, such as cetyltrimethylammonium bromide (CTAB).
Surfactant molecules are highly active components that progressively create more complex structures as concentration increases, while seeking a state of equilibrium or minimum energy [21]. At low concentrations, the surfactants exist as single molecules. By increasing their concentration in aqueous solutions, these molecules combine to form micelles to reduce the contact between the hydrophobic hydrocarbon chains and water, thus reducing the surface tension. In general, a silicon alkoxide promotes the formation of the silica structure outside the micelles and the presence of a catalyst facilitate the hydrolysis and condensation of silica precursors to form a network of siloxane bonds [43]. As an example, Figure 4 illustrates the formation of MCM-41. Initially, under a certain concentration, the surfactant molecules are free in the medium. Once their concentration increases and they reach the critical micelle concentration (CMC), the surfactants aggregate to form an isotropic micelle system. The concentration process continues and hexagonal assemblies appear close together, resulting in the formation of the hexagonal phases. The coalescence of adjacent, mutually parallel cylinders produces the lamellar phase. The formation of a particular phase depends not only on concentrations, but also on pH, temperature, ionic strength, solvent, the presence of other compounds, the nature of the surfactant itself (including the length of the hydrocarbon chain), the hydrophilic head group and the counter ion, in the case of ionic surfactants [23]. Finally, in order to incorporate payloads into the pores, the surfactant channels might be removed by calcination or via chemical treatment.
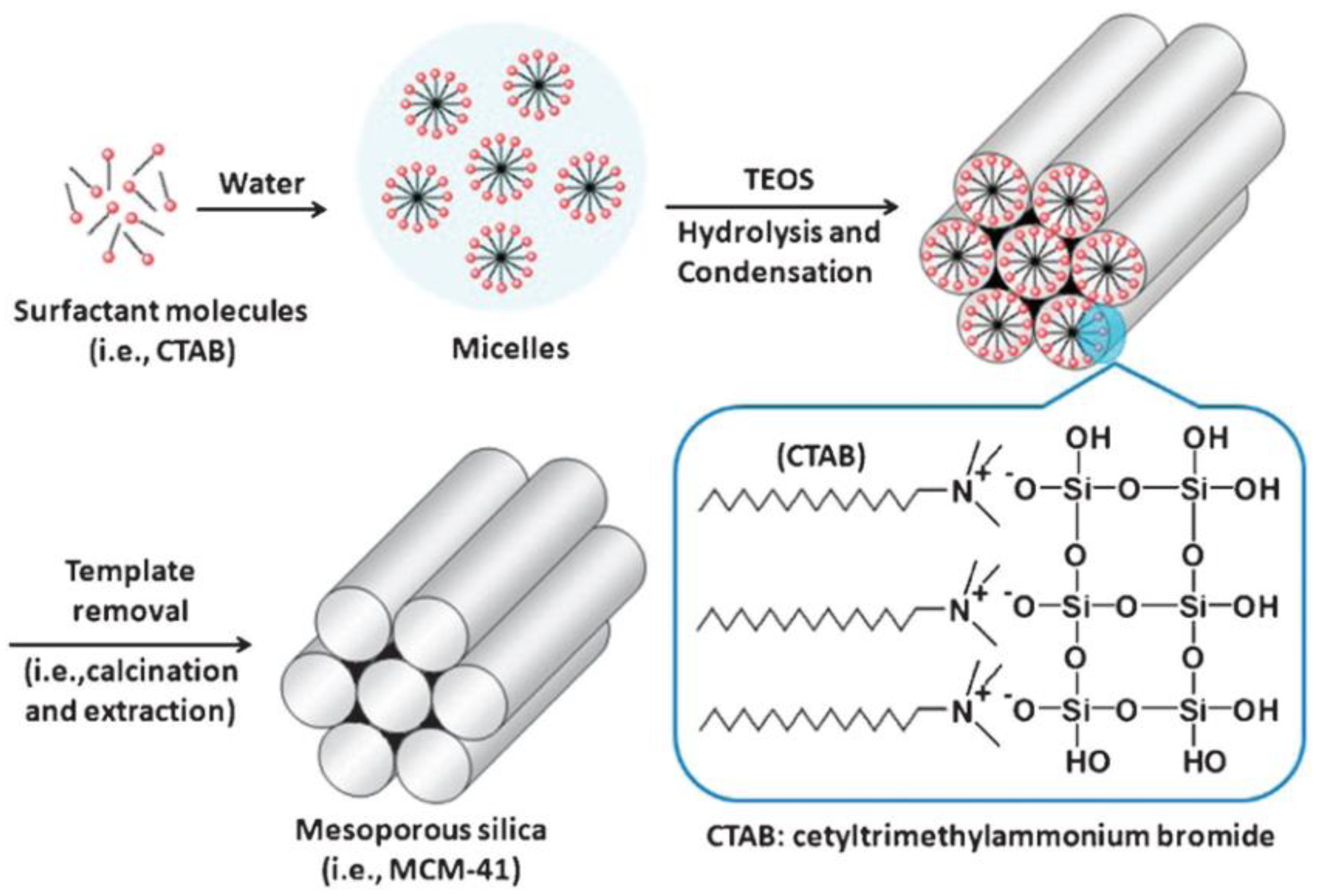
Figure 4. Schematic representation of the mechanism of formation of MCM-41 using cetyltrimethylammonium bromide (CTAB) as cationic surfactant.
Sol-Gel Process through Template-Assisted Technique
The template-assisted technique is the most popular formation mechanism for MCM-41, first postulated in 1992 by the Mobil scientists. According to this mechanism, surfactant molecules, such as alkyltrimethylammonium salts, form hexagonal arrays of micellar rods, which play a templating role (structure-directing agent). Afterwards, the silicate species assemble in between the surfactant tubules through the sol-gel process to form the inorganic framework [19][44]. Accordingly, the final product is a silicate skeleton that contains voids and mimics these mesophases [33]. Additionally, silica/surfactant mesophases are formed in the solution by one of two complementary approaches: (1) the cooperative assembly of small silicate species with micelles and individual surfactant molecules and (2) liquid crystal templating (LCT) of molecular inorganic species around a preformed, spatially extended organic superstructure [45][46]. The silicate species added to the reaction mixture may influence the ordering of the isotropic rod-like micelles to form the desired liquid crystal phase (hexagonal mesophase) [33]. On the other hand, the surfactant creates a hexagonal structure before the silica precipitates around this template, forming the mesoporous structure [18].
In fact, hexagonal ordering may be mediated by the interaction of silicate species with surfactants and both, the formation of surfactant liquid crystals and the assembly of silicate ions may occur simultaneously. It has been hypothesized that, during the formation of cylindrical surfactant/silica aggregates, silicate ions enter the particle by migrating from the surface toward the center of the particles (by a controlled diffusion process). Meanwhile, the surfactant molecules must enter through the cylinders of the aggregates, resulting in a continuous increase in organic content and channel diameter. This occurs more readily near the particle surface, where the network tension is lower than in the center [44].
Frasch and co-workers [47] proposed a similar model to the one reported by Cai et al. [48], in which silicate/surfactants rod-micelles form in the first step and then pack together in an ordered fashion to form the mesoporous nanoparticles [49].
Once the MSN structure has been obtained, removal of the surfactant or organic material is essential. This is usually achieved by calcination or solvent extraction. In the case of calcination, the synthesized MSNs are heated to temperatures between 300 °C and 600 °C [21][48]. However, at high temperatures, the particles can undergo surface modifications. Specifically, the Si-OH bonds on the surface of the MSNs react to form siloxane bonds. This constricts the surface and pores, modifying the pore size and making the particle more hydrophobic. Solvent extraction can also be used but the conditions will depend on the surfactant nature and whether the reaction occurred under acidic or basic conditions [21]. For the synthesis of mesoporous materials by sol-gel processes, different templates, such as cation surfactants, triblock copolymers and small organic molecules can be used as structure-directing agents [18].
Beside the sol-gel/template assisted synthesis of MSNs, the most widely investigated approaches are the microwave technology (which saves both energy and time, and the operation of the system is relatively simple) [50], hydrothermal and EISA. In all cases, it is necessary to introduce a templating agent to allow the formation of a nanoporous structure, in which the characteristics of the cavities (size, shape, orientation, etc.) are related to the configuration of the organic mold.
2.2.2. Factors Influencing the Size and Shape of Mesoporous Materials
The morphological and physicochemical properties of MSNs can be controlled by varying the synthesis conditions. MSNs are primarily formed in an ordered fashion from surfactants under acidic, basic or neutral conditions, which dictates their shape and size . MSNs can be synthesized using anionic, cationic, neutral surfactants or non-surfactant template pathways [51]. The diameter of the pores can be controlled by changing the length of the template molecule. Size, shape and porosity can be tuned by changing the silica source (e.g., colloidal silica or TEOS), surfactants (e.g., hexadecylamine (had) and cetyltrimethylammonium bromide (CTAB)), auxiliary compounds, or reaction conditions (e.g., solvent, temperature, time, stirring speed, reactant mole ratio and pH of the medium), thus affecting, at the same time, the properties and stability of the material [16][23].
Morphology control is extremely important for several applications. Not only have a variety of spherical or quasi-spherical MSNs with various porous and bulk properties been reported [42], but different morphologies, including thin films, hexagonal prisms, spirals, dodecahedrons and hollow tubular shapes of mesoporous silica, have also been synthesized. The main parameters for the synthesis of MSNs and their effects on particle properties are summarized in Figure 5.
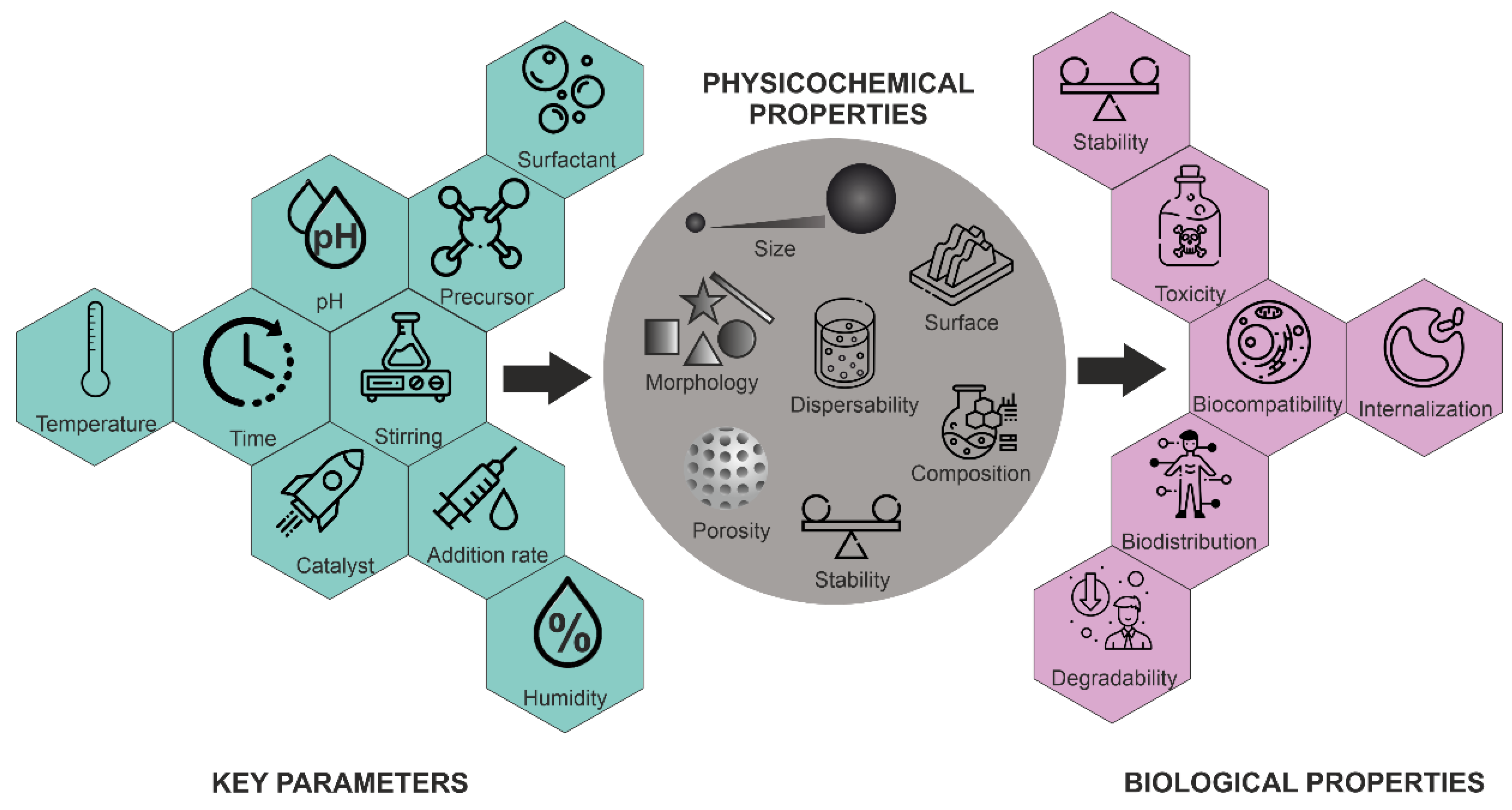
Figure 5. Influence of key parameters on the physico-chemical and biological properties of MSNs.
pH
The hydrolysis and condensation rates of silica species are strongly pH-dependent, which dictates both the reaction’s kinetics and the final network structure [23]. Therefore, pH is an important parameter and plays a critical role. It influences the synthetic chemistry of mesoporous materials, particularly affecting the charge of inorganic precursor species and surfactant head groups and, consequently, their mutual interaction [17]. At high pH values, the negatively charged inorganic precursors interact preferentially with the positively charged groups of the surfactants, resulting in condensation on the solid organic-inorganic mesoporous structures. In general, in a strongly acidic medium, the rate of the hydrolysis of TEOS is faster than the condensation. By changing the pH, a phase transformation of silica from the lamellar to the hexagonal phase has been reported [17]. The formation mechanism of mesophases under acidic and basic conditions is completely different because negatively charged silicate ions act as counter ions above the isoelectric point (pH > pI~2.0), while below the isoelectric point, positively charged silicate ions act as counter ions [17]. In addition, the dissociation constant (pKa) of the silanol groups (Si-OH)—which are present at the silica surface—has been determined in the range 4–5.5, indicating the dissociation of Si-OH groups, forming negatively charged Si-O− groups at the surface of silica [52].
As shown in Figure 6, at pH values lower than the isoelectric point (pH ~2), the condensation is acid-catalyzed [53], the silica species are also positively charged and the charge density increases as the pH decreases . At pH > 2, the condensation rate increases with a pH up to about 7.5 [53]. At this point, the silica species become negative and the charge density of silicates increases along with increasing pH, due to the favored nucleophilic attack. At pH ~2–7, silicates with a negative charge density tend to assemble with positively charged surfactants or neutral polymers through hydrogen bonds and electrostatic interactions. Under alkaline conditions (pH >7.5), silicates with a high negative charge density can only assemble with cationic surfactants by a single, but strong, electrostatic interaction. From this point on, the condensation rate reaches a maximum and decreases for pH >7.5, due to the gradual instability of the silicates at higher pH [20][54]. Thus, the condensation reaction of silicates can be represented as follows:
–Si–O− + HO–Si– → –Si–O–Si– + OH−
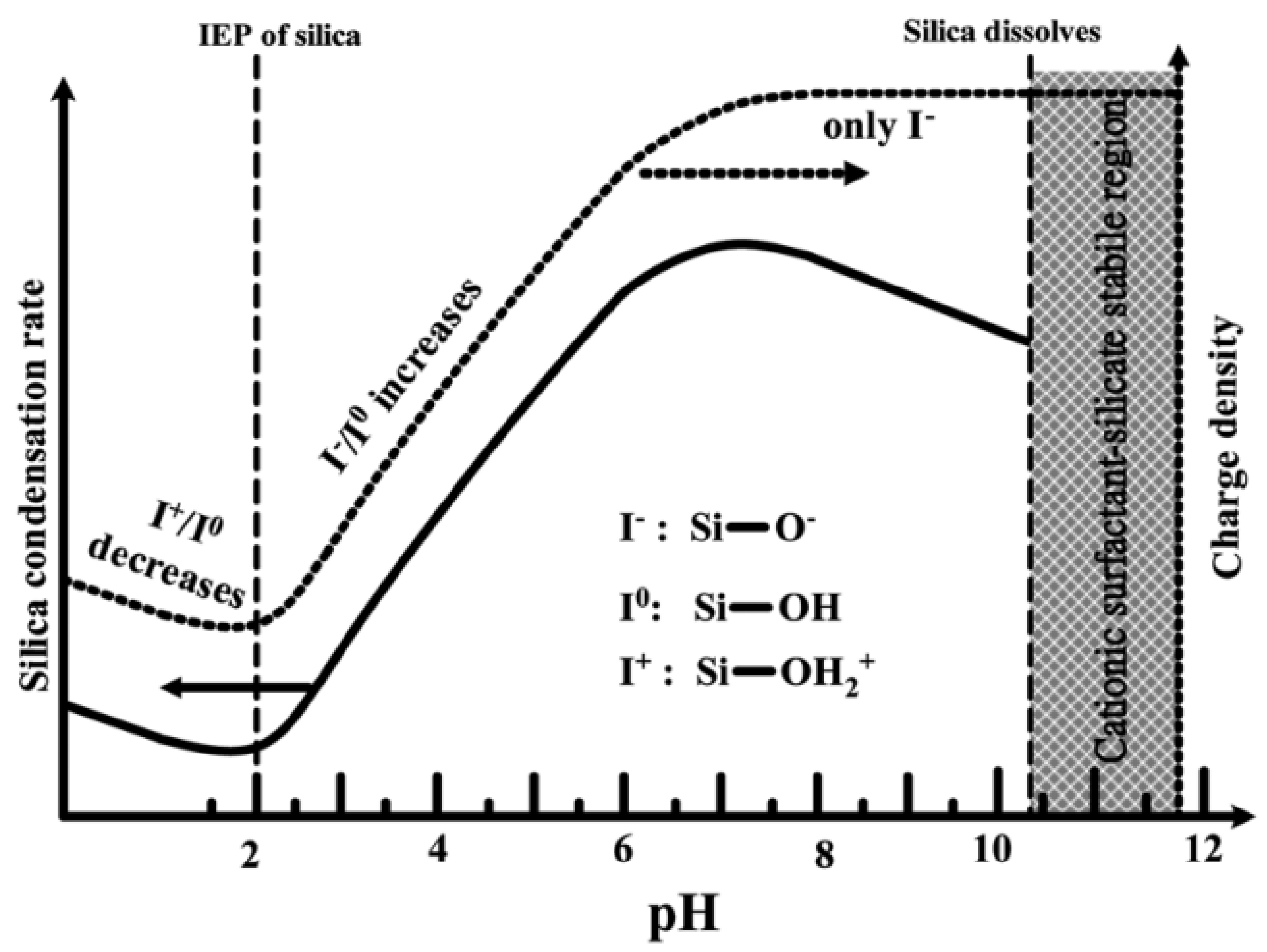
Figure 6. Effect of pH on the silica condensation rate and silica surface charge properties.
The effect of pH on the morphology of MSNs was studied by Yang et al. [55]. They demonstrated that, under mildly acidic conditions, 1–10 μm spherical mesoporous particles are formed. Furthermore, Voegtlin et al. [56] reported the room temperature synthesis of MSNs at pH values ranging from 8.5 to 12. Finally, Chiang et al. [57] studied all the parameters that can influence the particle size of MSNs, concluding that the pH played a key role [58].
Surfactant
Surfactant chemistry is critical for understanding the mesoporous structure formation. The pore size and structure, pore walls, or phase and symmetry of the materials are governed by the nature of the surfactant. Surfactant molecules have an amphiphilic nature, as they have a hydrophilic polar head group and a hydrophobic nonpolar hydrocarbon tail, being an active agent in an aqueous solution and showing high affinity to the surfaces and interfaces. Surfactants can be classified as anionic, cationic, zwitterionic and nonionic, according to the nature of their polar head group [17]. A homogenous spherical particle size distribution can be achieved at low concentration of CTAB (as cationic surfactant). Cai et al. [48] generated MSNs with different shapes, such as spheres, rods and micrometer-sized oblate silica by tuning the concentration of TEOS, NaOH/NH4OH and CTAB [16].
The pore width can also be adjusted, either during the synthesis or by post-synthesis hydrothermal treatments [59]. The pore size is mainly determined by the hydrophobic chain length of the surfactant. Different methods have been described to control pore size by the addition of organic auxiliary molecules or by using swelling agents, such as mesitylene, which solubilizes in the hydrophobic region, increasing the micellar size [54]. For instance, substituting the dodecyltrimethylammonium ion for a hexadecyltrimethylammonium ion (commonly known as CTAB) produced a sample of MCM-41 with 2–3 nm pore diameters [60]. Since additives can generate an imbalance in the reaction medium, post-synthesis hydrothermal treatment is an alternative to increase the pore size without inducing changes in the size and morphology of the preformed particles [59]. Furthermore, the use of co-solvent organic molecules, such as 1,3,5-trimethylbenzene (TMB), induces the formation of expanded pore sizes in MCM-41 [51].
Considering different modes of interaction between the silica precursors and surfactants, several models have been proposed [17]. Electrostatic charge-matching between cationic surfactants (S+) and anionic inorganic precursors (I−) is particularly effective for generating mesostructures with hexagonal, cubic or lamellar symmetry.
Schüth, Stucky and co-workers [61] included a charge-reversed S− I+ pathway between anionic surfactants, such as sulfonates, phosphonates, carboxylates and cationic precursors. Tanev and Pinnavaia [62] demonstrated that the assembly of hexagonal mesoporous metal oxides can be achieved between neutral amine surfactants (S0) and neutral inorganic precursors (I0) [63]. Hexagonal mesoporous silica with wormhole-like framework structures and larger wall thicknesses can be prepared by hydrogen bond and self-assembly between a neutral amine surfactants (S0) or nonionic PEO-based surfactant (N0) with neutral inorganic precursors (I0) [64].
Another approach for synthesizing small particles is the addition of an optimal amount of Pluronic F127 to the surfactant, which results in a change in the structure and packing of the micelles, leading to smaller particle sizes. In addition, Pluronic F127 also enhances the dispersion of the silica precursor (TEOS) and coats and stabilizes the newly formed small MSNs, helping to protect the particles from agglomeration and oligomerization [65].
Silica Source
Mesoporous structures have been synthesized using several silica sources, such as sodium silicate, Ludox, fumed-silica, water glass or silicon alkoxides (e.g., TEOS and TMOS), with the latter being the most widely reported [17]. Di Renzo and co-workers [66] described the synthesis of spherical MCM-41 using various silica sources, showing their dominating role in the morphology of MCM-41 silicates [67]. K. Yano and Y. Fukushima [68] succeeded in preparing large MSNs from alkoxysilanes with longer alkyl chain lengths, such as tetrapropoxysilane, since their hydrolysis rates are slower than those of tetramethoxysilane and tetraethoxysilane [30][69]. Nooney et al. [49] prepared MSNs with sizes ranging from 65 to 740 nm by using different TEOS/surfactant ratios under diluted conditions [58]. When the TEOS concentration increases, both the hydrolysis rate and condensation rate become faster, which shortens the nucleation period. Thus, the total number of nuclei formed will be less and the final particle size of synthetic silica colloids will be relatively larger.
Other Factors
Other factors that affect the size and shape of the MSNs include the temperature, amount and strength of the catalyst, reaction time and type of solvent. Thus, their combination should be considered to control the morphology of these particles [27]. The solvent is particularly important when silicon alkoxides are employed for the MSNs growth. Since these substances are nonpolar and, thus, insoluble in water, they are typically dissolved in a solution of water and ethanol [15], with the mixing mode having a significant effect on the homogeneity of the Si source in the reaction medium and its interaction with both the ethanol and water molecules [70]. Zhang et al. [13] reported that the dilution of TEOS with ethanol can improve the diffusion of TEOS in the mixture of ethanol and ammonia and depress the aggregation or adhesion of nanoparticles. It was found that four times the volume of ethanol for diluting TEOS is enough to obtain monodispersed nanoparticles with a fine spherical shape.
Depending on the type of solvent used in the reaction, different particle sizes and uniformity can be obtained. Stöber, Fink and Bohn [20] studied the particles’ growth using different alcohols as solvents. Particles prepared in methanol solutions turned out to be the smallest, while the particle size increased when increasing the length of the alcohol chain. The particle size distribution also became broader when using longer-chain alcohols as solvents [71], while more uniform particles were obtained in 1:3 mixtures of methanol/n-propanol. Additionally, Stöber et al. found that the size of the silica particles that can be obtained in TEOS/ethanol/water mixtures varied from 50 nm to about 1 µm.
The particle size increases with the concentration of catalyst (with ammonia being the most widely employed one for this purpose), as well as with an increase of TEOS [11]. Ammonia, as a morphogenic catalyst, leads to more spherical morphologies [49]. Cai et al. [48] synthesized different particle morphologies by selecting sodium hydroxide over ammonium hydroxide.
The final particle size is also strongly influenced by the reaction medium’s ionic strength and the catalyst and water concentrations. This is consistent with a proposed mechanism in which the final particle size was strongly dependent on the (intermediate) particle’s stability [72]. Nanoparticles prepared by the Stöber method usually have a relatively large amount of silanol groups, which allows the nanoparticles to disperse stably, due to their strong surface charges. When the solute concentration reaches a critical point (nucleation concentration), each solute self-assembles and forms small nanoparticles (thermodynamically favored). After nanoparticle formation, the solutes are also used for nanoparticle growth, and the nanoparticles gradually become larger. Accordingly, to reduce particle size, it is necessary to promote nucleation, rather than growth, by modulating the concentration of silica precursors [27].
As it has been mentioned before, the size of MSNs can be modified by the addition of certain compounds. Anderson et al. [46] reported the influence of various co-solvents and water mixtures on the size of MSNs. Gu et al. [73] proposed the use of EG, as it can decrease the interaction between the silica species and surfactants, thus reducing the size of nanoparticles [39][69]. Similarly, the addition of a second templating agent, with a different affinity for silica, can lead to the formation of a layer surrounding the particles, thus limiting their growth. The use of organo-substituted trialkoxysilane also plays a role in controlling MSN morphology and size. By using a series of alkyl substituted silanes, Huh et al. [74] generated diverse morphologies, such as rods, beans, or spheres, with precisely controlled particle sizes. [59]. Schulz-Ekloff et al. [36] described a method for obtaining materials of a novel type, namely bimodal silicas, which contain both the MCM-41 mesopore systems, with pore sizes ranging from about 2 to 15 nm [39][55][75].
Temperature has a greater effect on the rate of hydrolysis than other factors, such as co-solvents. Therefore, a relatively high temperature, such as 80 °C, is essential when using tetraalkoxysilanes with slow hydrolysis rates [76]. As a consequence, the reaction temperature can change the properties of the particles, mainly affecting the particle size. For example, Narayan, Reema et al. [16] observed that, by increasing the temperature of the reaction from 30 to 70 °C, particle size was increased from ca. 29 nm to 113 nm. This could be due to the increase of the reaction rate, leading to polycondensation of the silica monomers and resulting in a dense silica structure and a larger size.
In summary, the parameters that control the size and morphology of MSNs include the rate of hydrolysis and condensation (pH-dependent) and the level of interaction between the assembled template and the silica precursor [58][59][77].
2.3. Hollow Silica Nanoparticles
Hollow spheres represent a special class of materials that are of interest, for instance, in the fields of medicine, pharmaceutics, materials science, catalysis and the paint industry. They can encapsulate a variety of products (for the controlled release of drugs, cosmetics, inks and dyes), protect light-sensitive compounds and to be employed in coatings, composites and fillers [75]. Hollow silica nanoparticles (HSNs) refer to NPs with a solid shell, even though a recent trend has been to synthesize hollow nanospheres with a mesoporous silica shell. An advantage of the hollow form of MSNs is that their inner cavities can be used as containers for other cargo, such as magnetic, gold, or even smaller silica nanoparticles [78]. Hollow MSNs (HMSNs), a sub-class of MSNs with interstitial hollow space with extraordinarily high loading capacity, low density and high specific area, have been advocated as the new-generation of DDSs [58]. They are capable of holding a large amount of drugs in their hollow cavities, compared to their non-hollow counterparts. This unique property of HMSNs makes them widely useful in cancer therapy and imaging [16].
The development of novel fabrication methods for hollow MSNs has been one of the most active areas of research in nanotechnology. Numerous fabrication techniques exist to obtain hollow spheres with a wide range of diameters and wall thicknesses [75], including microemulsions, soft templating and hard templating [15]. However, the most frequently used approach for the synthesis of HMSNs is the core-templating method, where different soft and hard templates are used to form the nanostructure core that is then coated with the desired substance at different concentrations to obtain a shell around the substrate. After that, the template forming the core is removed by calcination or chemical etching, obtaining a shell with a hollow core [16]. Choosing the appropriate core template is critical for producing efficient hollow capsules, as their properties, such as size, shape and ease of removal, determine the properties of the final hollow capsules. Hollow nanocapsules with various diameters can, thus, be prepared by changing the diameter of these templates [79].
Hollow-type mesopores are created in the core-templating technique by exploiting the chemical differences between the core and shell of a silica core/mesoporous silica structure, in such a way that the core can be removed by heating (calcination) or by dissolution [75]. When an appropriate etching agent is used, a selective etching takes place at the interior, while the outer shell remains mostly intact and the hollow structure is formed [18]. The porous structure on the surface of the silica NPs provides a pathway for the etchant to penetrate the core of the silica material, thus facilitating further etching. Furthermore, when the silicate species reach the supersaturation level at the interface, condensation is expected to occur, leading to the expansion of the inner shell, while maintaining the overall shape [80]. There are some soft templates based on aggregates of organic molecules, including large micelles, vesicles and emulsions. Their structure and morphology can be easily controlled with the change of the preparative conditions, which enables various hollow-structured MSNs to be obtained. In the final step, they can be easily removed through calcination. Therefore, many hollow-structured MSNs have been prepared by using soft templates [78]. Tanev and Pinnavaia [81] condensed silica in the interlayer regions of multilamellar vesicles to form roughly spherical particles with stable lamellar mesostructures [82]. Using another approach, Lin et al. [83] prepared thermally stable NPs in a water-in-oil (w/o) emulsion, consisting of water, hydrocarbons and cationic surfactants [58].
The hard template method hollow-structured MSNs can be obtained with the selective elimination of the core particles [27]. Polymeric nanoparticles and carbon nanoparticles are some of the most frequently used hard templates, but metal oxide, metal nanoparticles and other inorganic nanoparticles have also been employed. Le et al. [42] described a synthesis pathway to obtain hollow silica spheres (40 nm average diameter) with mesoporous walls. The process was based on a double-template route using calcium carbonate nanoparticles as sphere templates and hexadecyltrimethylammonium bromide (C16TMABr) as a mesostructure-directing agent in alkaline conditions, obtaining silica hollow spheres at room temperature. The use of silica nanoparticles as templates (core particles) and coated with mesoporous silica shells has been widely reported. These silica cores present highly tunable characteristics, such as diameter, morphology and size distribution. The major problem of silica core nanoparticles is that separating the core from the silica shell requires a sophisticated procedure for selective core etching. For example, hollow organosiloxane NPs can be obtained by selectively etching the silica cores, thus exploiting the tolerance of organosiloxane toward base etching [27].
In the biomedical field, both discrete and monodispersed HMSNs play key roles in providing enough stability to the cargo in physiological environments and their nano-sizes enable effective distribution of the drugs in the body. Moreover, due to their hollow cavities, HMSNs have a large capacity to load biomedicines, enzymes, or small nanoparticles [58].
2.4. Other Silica Nanoparticles
While the development of silica NPs has historically been focused on MSNs, two additional types, mesoporous organosilica nanoparticles (MONs) and periodic mesoporous organosilica (PMO) NPs, are budding sectors in current silica NPs’ development, being one of the most representative inorganic–organic hybrid mesoporous nanoparticles. While PMO NPs and MONs are quite new, they already account for at least 10% of the papers currently being published on silica NPs [21].
As mentioned, when a single silica precursor, such as TEOS, is used, MSNs are formed. MONs will form by inserting organic groups into the framework of MSNs at the molecular level. The use of 100% of bridget-organosilane precursors, when no additional silane source is present, leads to the formation of PMO NPs [84][85]. Therefore, MONs are partially hybridized silica NPs, while PMO are exclusively formed by organically modified silicon atoms. This allows for the formation of a wide variety of additional silica nanoparticles with new chemistries, due to the presence of the organic (R) group.
As for MSNs, the most employed method is the sol-gel synthesis under basic or acid conditions, using surfactants to obtain the porous structure. When silanes and organosilanes ([(XO)3Si]n–R, where R is an organic group, n ≥ 1) are hydrolyzed in basic media, reactive silanolate species are produced. These species then condense with other (organo)silanes to create covalent siloxane linkages and progressively bigger oligomers. The sol-gel process eventually results in silica (SiO2) or silsesquioxane frameworks (e.g., O1.5Si-R-SiO1.5). Although some organically bridged structures are stable up to 580 °C, calcination is not likely to be a good alternative for MONs and PMO NPs, which is the method most commonly used to remove the surfactants from MSNs. Therefore, less aggressive extraction procedures are required [84].
MONs and PMO structures provide exponentially more design possibilities, as compared with MSNs, with the high potential of making uniquely functional silica nanoparticles, due to the flexibility of the organic R group, as previously mentioned [15]. Homogeneously distributed organic moieties throughout the mesoporous framework have a crucial impact on the size, the porosity and the morphology of the hybrid nanomaterials [86][87]. The R group can produce both physical changes to the pore size and shape and chemical changes affecting the hydrophobicity/hydrophilicity and charge of the particle have an impact on their final applications, such as the type of cargo that can be loaded on them or their loading capacities [21][88]. To add additional complexity, MONs have regions of MSNs and PMO that create hybrids, which can have different proportions of the organic and non-organic components, leading to the formation of unique porous structures [21]. Importantly, modifications of the R group allow for control over the biodegradability of these NPs, being a key point for their biomedical application [85][89].
MONs and PMO NPs have been advocated as some of the most prosperous nanomaterials over the next decade for their application in multiple fields, such as catalysis, gas and molecule adsorption, electronics, drug and gene delivery, biosensing, photodynamic therapy, or molecular imaging, among others [21][88].
Despite the benefits that MON and PMO NPs offer, their use in drug delivery has not received as much research attention as that of MSNs of pure silica. This could account for the difficulty in synthesizing uniform and discrete NPs with adjustable properties and the lack of knowledge regarding their biosafety [14][90]. In addition, despite the potential of MONs and PMO NPs for medical applications, they will encounter a larger barrier than MSNs to reach the clinic. This is because toxicological studies on MSNs do not translate over to MONs or PMOs, and each R group arrangement would require further toxicological testing. Thus, although FDA considers silica as safe, MONs and PMO NPs are not benefited by this because they are regarded as brand new compounds [21][84].
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14122703
References
- Zhao, P.; Liu, M.-C.; Lin, H.-C.; Sun, X.-Y.; Li, Y.-Y.; Yan, S.-Q. Synthesis and Drug Delivery Applications for Mesoporous Silica Nanoparticles. J. Med. Biotechnol. 2017, 1, 47.
- Deepak, T.; Michel, D.; Yashwant, P. Nanoparticulate Drug-Delivery Systems: An Overview; CRC Press: Boca Raton, FL, USA, 2007; Chapter 1; pp. 1–32.
- Rahman, I.A.; Padavettan, V. Synthesis of Silica Nanoparticles by Sol-Gel: Size-Dependent Properties, Surface Modification, and Applications in Silica-Polymer Nanocompositesa Review. J. Nanomater. 2012, 2012, 1–15.
- Meier, W. Nanostructure Synthesis Using Surfactants and Copolymers. Curr. Opin. Colloid Interface Sci. 1999, 4, 6–14.
- Fowler, C.E.; Khushalani, D.; Lebeau, B.; Mann, S. Nanoscale Materials with Mesostructured Interiors. Adv. Mater. 2001, 13, 649–652.
- Asefa, T.; Tao, Z. Biocompatibility of Mesoporous Silica Nanoparticles. Chem. Res. Toxicol. 2012, 25, 2265–2284.
- Jeelani, P.G.; Mulay, P.; Venkat, R.; Ramalingam, C. Multifaceted Application of Silica Nanoparticles. A Review. Silicon 2020, 12, 1337–1354.
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Flatt, R.J.; Estrela-Lopis, I.; Llop, J.; Moya, S.; et al. Nanoparticle Decoration with Surfactants: Molecular Interactions, Assembly, and Applications. Surf. Sci. Rep. 2017, 72, 1–58.
- Morales, M.E.; Castán, H.; Ortega, E.; Ruiz, M.A. Silica Nanoparticles: Preparation, Characterization and Applications in Biomedicine. Pharm. Chem. J. 2019, 53, 329–336.
- Lin, Y.S.; Haynes, C.L. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. J. Am. Chem. Soc. 2010, 132, 4834–4842.
- Ibrahim, I.A.; Zikry, A.A.F.; Sharaf, M.A. Preparation of Spherical Silica Nanoparticles: Stober Silica. J. Am. Sci. 2010, 6, 985–989.
- Graf, C. Silica, Amorphous. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley & Sons: New York, NY, USA, 2018; Volume 3, ISBN 0471238961.
- Zhang, J.H.; Zhan, P.; Wang, Z.L.; Zhang, W.Y.; Ming, N.B. Preparation of Monodisperse Silica Particles with Controllable Size and Shape. J. Mater. Res. 2003, 18, 649–653.
- Vallet-Regí, M.; Schüth, F.; Lozano, D.; Colilla, M.; Manzano, M. Engineering Mesoporous Silica Nanoparticles for Drug Delivery: Where Are We after Two Decades? Chem. Soc. Rev. 2022, 51, 5365–5451.
- Tang, F.; Li, L.; Chen, D. Mesoporous Silica Nanoparticles: Synthesis, Biocompatibility and Drug Delivery. Adv. Mater. 2012, 24, 1504–1534.
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118.
- Naik, B.; Ghosh, N. A Review on Chemical Methodologies for Preparation of Mesoporous Silica and Alumina Based Materials. Recent Pat. Nanotechnol. 2009, 3, 213–224.
- Kumar, S.; Malik, M.M.; Purohit, R. Synthesis Methods of Mesoporous Silica Materials. Mater. Today Proc. 2017, 4, 350–357.
- Kolbe, G. Das Komplexechemische Verhalten Der Kieselsäure. Ph.D. Thesis, Friedrich-Schiller-Universität, Jena, Germany, 1956.
- Stöber, W.; Fink, A.; Bohn, E. Controlled Growth of Monodisperse Silica Spheres in the Micron Size Range. J. Colloid Interface Sci. 1968, 26, 62–69.
- Downing, M.A.; Jain, P.K. Mesoporous Silica Nanoparticles: Synthesis, Properties, and Biomedical Applications; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128166628.
- Díaz de Greñu, B.; de los Reyes, R.; Costero, A.M.; Amorós, P.; Ros-Lis, J.V. Recent Progress of Microwave-Assisted Synthesis of Silica Materials. Nanomaterials 2020, 10, 1092.
- ALOthman, Z.A. A Review: Fundamental Aspects of Silicate Mesoporous Materials. Materials 2012, 5, 2874–2902.
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I. Mesoporous Silica Nanoparticles in Biomedical Applications. Chem. Soc. Rev. 2012, 41, 2590–2605.
- Lee, M.H.; Beyer, F.L.; Furst, E.M. Synthesis of Monodisperse Fluorescent Core-Shell Silica Particles Using a Modified Stöber Method for Imaging Individual Particles in Dense Colloidal Suspensions. J. Colloid Interface Sci. 2005, 288, 114–123.
- Finnie, K.S.; Bartlett, J.R.; Barbe, C.J.A.; Kong, L. Formation of Silica Nanoparticles in Microemulsions. Langmuir 2007, 23, 3017–3024.
- Yamamoto, E.; Kuroda, K. Colloidal Mesoporous Silica Nanoparticles. Bull. Chem. Soc. Jpn. 2016, 89, 501–539.
- Peres, E.C.; Slaviero, J.C.; Cunha, A.M.; Hosseini–Bandegharaei, A.; Dotto, G.L. Microwave Synthesis of Silica Nanoparticles and Its Application for Methylene Blue Adsorption. J. Environ. Chem. Eng. 2018, 6, 649–659.
- Lovingood, D.D.; Owens, J.R.; Seeber, M.; Kornev, K.G.; Luzinov, I. Controlled Microwave-Assisted Growth of Silica Nanoparticles under Acid Catalysis. ACS Appl. Mater. Interfaces 2012, 4, 6875–6883.
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41.
- Øye, G.; Sjöblom, J.; Stöcker, M. Synthesis, Characterization and Potential Applications of New Materials in the Mesoporous Range. Adv. Colloid Interface Sci. 2001, 89–90, 439–466.
- Yanagisawa, T.; Shimizu, T.; Kuroda, K.; Kato, C. The Preparation of Alkyltrimethylammonium-Kanemite Complexes and Their Conversion to Microporous Materials. Bull. Chem. Soc. Jpn. 1990, 63, 988–992.
- Zhao, X.S.; Lu, G.Q.; Millar, G.J. Advances in Mesoporous Molecular Sieve MCM-41. Ind. Eng. Chem. Res. 1996, 35, 2075–2090.
- Schulz-Ekloff, G.; Rathouský, J.; Zukal, A. Mesoporous Silica with Controlled Porous Structure and Regular Morphology. Int. J. Inorg. Mater. 1999, 1, 97–102.
- Grün, M.; Unger, K.K.; Matsumoto, A.; Tsutsumi, K. Novel Pathways for the Preparation of Mesoporous MCM-41 Materials: Control of Porosity and Morphology. Microporous Mesoporous Mater. 1999, 27, 207–216.
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.-T.; Lin, V.S.-Y. Synthesis and Functionalization of a Mesoporous Silica Nanoparticle Based on the Sol–Gel Process and Applications in Controlled Release. Acc. Chem. Res. 2007, 40, 846–853.
- Hoang Thi, T.T.; Cao, V.D.; Nguyen, T.N.Q.; Hoang, D.T.; Ngo, V.C.; Nguyen, D.H. Functionalized Mesoporous Silica Nanoparticles and Biomedical Applications. Mater. Sci. Eng. C 2019, 99, 631–656.
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.Y. Mesoporous Silica Nanoparticles for Drug Delivery and Biosensing Applications. Adv. Funct. Mater. 2007, 17, 1225–1236.
- Wu, K.C.W.; Yamauchi, Y. Controlling Physical Features of Mesoporous Silica Nanoparticles (MSNs) for Emerging Applications. J. Mater. Chem. 2012, 22, 1251–1256.
- Colilla, M.; González, B.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for the Design of Smart Delivery Nanodevices. Biomater. Sci. 2013, 1, 114–134.
- Grün, M.; Lauer, I.; Unger, K.K. The Synthesis of Micrometer- and Submicrometer-Size Spheres of Ordered Mesoporous Oxide MCM-41. Adv. Mater. 1997, 9, 254–257.
- Le, Y.; Chen, J.F.; Wang, J.X.; Shao, L.; Wang, W.C. A Novel Pathway for Synthesis of Silica Hollow Spheres with Mesostructured Walls. Mater. Lett. 2004, 58, 2105–2108.
- Rastegari, E.; Hsiao, Y.-J.; Lai, W.-Y.; Lai, Y.-H.; Yang, T.-C.; Chen, S.-J.; Huang, P.-I.; Chiou, S.-H.; Mou, C.-Y.; Chien, Y. An Update on Mesoporous Silica Nanoparticle Applications in Nanomedicine. Pharmaceutics 2021, 13, 1067.
- Zhou, W. HRTEM Investigation of Mesoporous Molecular Sieves. Micron 2000, 31, 605–611.
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843.
- Anderson, M.T.; Martin, J.E.; Odinek, J.G.; Newcomer, P.P. Surfactant-Templated Silica Mesophases Formed in Water: Cosolvent Mixtures. Chem. Mater. 1998, 10, 311–321.
- Zana, R.; Frasch, J.; Soulard, M.; Lebeau, B.; Patarin, J. Fluorescence Probing Investigations of the Mechanism of Formation of Organized Mesoporous Silica. Langmuir 1999, 15, 2603–2606.
- Cai, Q.; Luo, Z.S.; Pang, W.Q.; Fan, Y.W.; Chen, X.H.; Cui, F.Z. Dilute Solution Routes to Various Controllable Morphologies of MCM-41 Silica with a Basic Medium. Chem. Mater. 2001, 13, 258–263.
- Nooney, R.I.; Thirunavukkarasu, D.; Yimei, C.; Josephs, R.; Ostafin, A.E. Synthesis of Nanoscale Mesoporous Silica Spheres with Controlled Particle Size. Chem. Mater. 2002, 14, 4721–4728.
- Wu, C.-G.; Bein, T. Microwave Synthesis of Molecular Sieve MCM-41. Chem. Commun. 1996, 8, 925–926.
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores. Science 1998, 279, 548–552.
- Allen, L.H.; Matijevíc, E.; Meites, L. Exchange of Na+ for the Silanolic Protons of Silica. J. Inorg. Nucl. Chem. 1971, 33, 1293–1299.
- Lin, H.P.; Mou, C.Y. Structural and Morphological Control of Cationic Surfactant-Templated Mesoporous Silica. Acc. Chem. Res. 2002, 35, 927–935.
- Vallet-Regi, M.; Rámila, A.; Del Real, R.P.; Pérez-Pariente, J. A New Property of MCM-41: Drug Delivery System. Chem. Mater. 2001, 13, 308–311.
- Yang, H.; Vovk, G.; Coombs, N.; Sokolov, I.; Ozin, G.A. Synthesis of Mesoporous Silica Spheres under Quiescent Aqueous Acidic Conditions. J. Mater. Chem. 1998, 8, 743–750.
- Voegtlin, A.C.; Matijasic, A.; Patarin, J.; Sauerland, C.; Grillet, Y.; Huve, L. Room-Temperature Synthesis of Silicate Mesoporous MCM-41-Type Materials: Influence of the Synthesis PH on the Porosity of the Materials Obtained. Microporous Mater. 1997, 10, 137–147.
- Chiang, Y.-D.; Lian, H.-Y.; Leo, S.-Y.; Wang, S.-G.; Yamauchi, Y.; Wu, K.C.-W. Controlling Particle Size and Structural Properties of Mesoporous Silica Nanoparticles Using the Taguchi Method. J. Phys. Chem. C 2011, 115, 13158–13165.
- Mehmood, A.; Ghafar, H.; Yaqoob, S.; Gohar, U.F.; Ahmad, B. Mesoporous Silica Nanoparticles: A Review. J. Dev. Drugs 2017, 6.
- Slowing, I.I.; Vivero-Escoto, J.L.; Trewyn, B.G.; Lin, V.S.Y. Mesoporous Silica Nanoparticles: Structural Design and Applications. J. Mater. Chem. 2010, 20, 7924–7937.
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism. Nature 1992, 359, 710–712.
- Huo, Q.; Margolese, D.I.; Ciesla, U.; Feng, P.; Gier, T.E.; Sieger, P.; Leon, R.; Petroff, P.M.; Schüth, F.; Stucky, G.D. Generalized Synthesis of Periodic Surfactant/Inorganic Composite Materials. Nature 1994, 368, 317–321.
- Tanev, P.T.; Pinnavaia, T.J. A Neutral Templating Route to Mesoporous Molecular Sieves. Science 1995, 267, 865–867.
- Bagshaw, S.A.; Prouzet, E.; Pinnavaia, T.J. Templating of Mesoporous Molecular Sieves by Nonionic Polyethylene Oxide Surfactants. Science 1995, 269, 1242–1244.
- Lin, H.Y.; Chen, Y.W. Preparation of Spherical Hexagonal Mesoporous Silica. J. Porous Mater. 2005, 12, 95–105.
- Mai, W.X.; Meng, H. Mesoporous Silica Nanoparticles: A Multifunctional Nano Therapeutic System. Integr. Biol. 2013, 5, 19–28.
- Martin, T.; Galarneau, A.; Di Renzo, F.; Fajula, F.; Plee, D. Morphological Control of MCM-41 by Pseudomorphic Synthesis. Angew. Chem. Int. Ed. 2002, 41, 2590–2592.
- Szegedi, Á.; Kónya, Z.; Méhn, D.; Solymár, E.; Pál-Borbély, G.; Horváth, Z.E.; Biró, L.P.; Kiricsi, I. Spherical Mesoporous MCM-41 Materials Containing Transition Metals: Synthesis and Characterization. Appl. Catal. A Gen. 2004, 272, 257–266.
- Yano, K.; Fukushima, Y. Synthesis of Mono-Dispersed Mesoporous Silica Spheres with Highly Ordered Hexagonal Regularity Using Conventional Alkyltrimethylammonium Halide as a Surfactant. J. Mater. Chem. 2004, 14, 1579–1584.
- Nandiyanto, A.B.D.; Kim, S.-G.; Iskandar, F.; Okuyama, K. Synthesis of Spherical Mesoporous Silica Nanoparticles with Nanometer-Size Controllable Pores and Outer Diameters. Microporous Mesoporous Mater. 2009, 120, 447–453.
- Jafarzadeh, M.; Rahman, I.A.; Sipaut, C.S. Synthesis of Silica Nanoparticles by Modified Sol-Gel Process: The Effect of Mixing Modes of the Reactants and Drying Techniques. J. Sol-Gel Sci. Technol. 2009, 50, 328–336.
- Lindberg, R.; Sjöblom, J.; Sundholm, G. Preparation of Silica Particles Utilizing the Sol-Gel and the Emulsion-Gel Processes. Colloids Surf. A Physicochem. Eng. Asp. 1995, 99, 79–88.
- Van Blaaderen, A.; Van Geest, J.; Vrij, A. Monodisperse Colloidal Silica Spheres from Tetraalkoxysilanes: Particle Formation and Growth Mechanism. J. Colloid Interface Sci. 1992, 154, 481–501.
- Gu, J.; Fan, W.; Shimojima, A.; Okubo, T. Organic-Inorganic Mesoporous Nanocarriers Integrated with Biogenic Ligands. Small 2007, 3, 1740–1744.
- Huh, S.; Wiench, J.W.; Yoo, J.-C.; Pruski, M.; Lin, V.S.-Y. Organic Functionalization and Morphology Control of Mesoporous Silicas via a Co-Condensation Synthesis Method. Chem. Mater. 2003, 15, 4247–4256.
- Caruso, F.; Caruso, R.A.; Möhwald, H. Production of Hollow Microspheres from Nanostructured Composite Particles. Chem. Mater. 1999, 11, 3309–3314.
- Yamada, H.; Urata, C.; Aoyama, Y.; Osada, S.; Yamauchi, Y.; Kuroda, K. Preparation of Colloidal Mesoporous Silica Nanoparticles with Different Diameters and Their Unique Degradation Behavior in Static Aqueous Systems. Chem. Mater. 2012, 24, 1462–1471.
- He, Q.; Cui, X.; Cui, F.; Guo, L.; Shi, J. Size-Controlled Synthesis of Monodispersed Mesoporous Silica Nano-Spheres under a Neutral Condition. Microporous Mesoporous Mater. 2009, 117, 609–616.
- Wu, S.H.; Hung, Y.; Mou, C.Y. Mesoporous Silica Nanoparticles as Nanocarriers. Chem. Commun. 2011, 47, 9972–9985.
- Itoh, Y.; Matsusaki, M.; Kida, T.; Akashi, M. Preparation of Biodegradable Hollow Nanocapsules by Silica Template Method. Chem. Lett. 2004, 33, 1552–1553.
- Zhang, H.; Zhou, Y.; Li, Y.; Bandosz, T.J.; Akins, D.L. Synthesis of Hollow Ellipsoidal Silica Nanostructures Using a Wet-Chemical Etching Approach. J. Colloid Interface Sci. 2012, 375, 106–111.
- Tanev, P.T.; Pinnavaia, T.J. Biomimetic Templating of Porous Lamellar Silicas by Vesicular Surfactant Assemblies. Science 1996, 271, 1267–1269.
- Li, W.; Coppens, M. Synthesis and Characterization of Stable Hollow Ti-Silica. Chem. Mater. 2005, 17, 2241–2246.
- Lin, Y.S.; Wu, S.H.; Tseng, C.T.; Hung, Y.; Chang, C.; Mou, C.Y. Synthesis of Hollow Silica Nanospheres with a Microemulsion as the Template. Chem. Commun. 2009, 24, 3542–3544.
- Croissant, J.G.; Fatieiev, Y.; Almalik, A.; Khashab, N.M. Mesoporous Silica and Organosilica Nanoparticles: Physical Chemistry, Biosafety, Delivery Strategies, and Biomedical Applications. Adv. Healthc. Mater. 2018, 7, 1700831.
- Poscher, V.; Salinas, Y. Trends in Degradable Mesoporous Organosilica-Based Nanomaterials for Controlling Drug Delivery: A Mini Review. Materials 2020, 13, 3668.
- Díaz Morales, U.M.; Corma Canós, A. Organic-Inorganic Hybrid Materials: MultiFunctional Solids for Multi-Step Reaction Processes. Chem.—A Eur. J. 2018, 24, 3944–3958.
- Erigoni, A.; Diaz, U. Porous Silica-Based Organic-Inorganic Hybrid Catalysts: A Review. Catalysts 2021, 11, 79.
- Croissant, J.G.; Cattoën, X.; Wong Chi Man, M.; Durand, J.-O.; Khashab, N.M. Syntheses and Applications of Periodic Mesoporous Organosilica Nanoparticles. Nanoscale 2015, 7, 20318–20334.
- Chinnathambi, S.; Tamanoi, F. Recent Development to Explore the Use of Biodegradable Periodic Mesoporous Organosilica (BPMO) Nanomaterials for Cancer Therapy. Pharmaceutics 2020, 12, 890.
- Yu, L.; Chen, Y.; Lin, H.; Du, W.; Chen, H.; Shi, J. Ultrasmall Mesoporous Organosilica Nanoparticles: Morphology Modulations and Redox-Responsive Biodegradability for Tumor-Specific Drug Delivery. Biomaterials 2018, 161, 292–305.
This entry is offline, you can click here to edit this entry!