Based on the literature data from 1973 to 2022, this work summarizes reports on spiro-flavonoids with a spiro-carbon at the center of their structure and how this affects their isolation methods, stereochemistry, and biological activity. The review collects 65 unique structures, including spiro-biflavonoids, spiro-triflavonoids, spiro-tetraflavonoids, spiro-flavostilbenoids, and scillascillin-type homoisoflavonoids. Scillascillin-type homoisoflavonoids comprise spiro[bicyclo[4.2.0]octane-7,3′-chromane]-1(6),2,4-trien-4′-one, while the other spiro-flavonoids contain either 2H,2′H-3,3′-spirobi[benzofuran]-2-one or 2′H,3H-2,3′-spirobi[benzofuran]-3-one in the core of their structures. Spiro-flavonoids have been described in more than 40 species of eight families, including Asparagaceae, Cistaceae, Cupressaceae, Fabaceae, Pentaphylacaceae, Pinaceae, Thymelaeaceae, and Vitaceae. The possible biosynthetic pathways for each group of spiro-flavonoids are summarized in detail. Anti-inflammatory and anticancer activities are the most important biological activities of spiro-flavonoids, both in vitro and in vivo. Our work identifies the most promising natural sources, the existing challenges in assigning the stereochemistry of these compounds, and future research perspectives.
- spiro-flavonoids
- spiro-biflavonoids
- spiro-triflavonoids
- spiro-tetraflavonoids
- spiro-flavostilbenoids
- scillascillin-type homoisoflavonoids
- biosynthesis
- biological activity
- isolation
1. Introduction
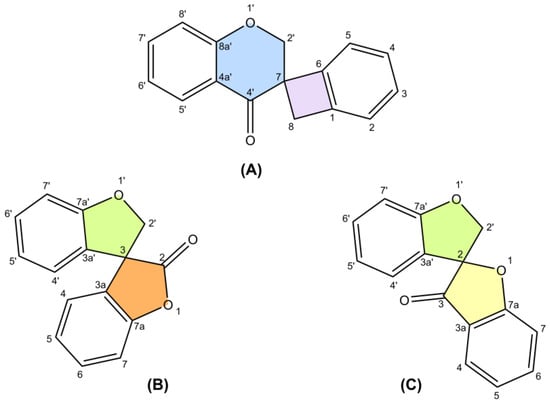
The only review paper on spiro-flavonoids was published by Fedorova et al. [8] and covered eight spiro-biflavonoids and one spiro-triflavonoid that had been isolated prior to 2010, without any discussion of their stereochemistry. Thanks to modern and increasingly available spectroscopic techniques, more than 50 additional spiro-flavonoids have been characterized to date, resulting in a total of 65 unique structures, and are reported here. Difficulties in assigning stereochemistry, artifacts and discovering spiro-flavonoids already described under another name were discussed.
This review summarizes the literature data for the years 1973-2022, including the authors' studies, on the occurrence in nature, methods of isolation, elucidation of structures, assignment of stereochemistry, bioactivity, and putative biosynthesis of spiro-flavonoids.
POWO "Plants of the World Online" [9] and IPNI "International Plant Names Index" [10] were used to verify the plant and family names (Table 1). Common numbering of carbon atoms of spiro compounds was used according to Nakashima et al. (2016) and Pecio et al. (2023) [7,11].
2. Occurrence in Nature
Spiro-flavonoids are a fairly large group of polyphenolic compounds (although it is less numerous by an order of magnitude than biflavonoids [12]), and to our knowledge a total of 65 have been described, including spiro-biflavonoids, spiro-triflavonoids, spiro-tetraflavonoids, spiro-flavostilbenoids, and scillascillin-type homoisoflavonoids (Tables 2-6). These first four groups of compounds reported here are composed of two or more nonidentical flavonoid and/or stilbene units, whereas homoisoflavonoids, with one exception, are monomeric structures. Interestingly, to date, no glycosylated forms of spiro-flavonoids have been described. Spiro-biflavonoids are the most abundant group, followed by scillascillin-type homoisoflavonoids and spiro-flavostilbenoids (29, 17 and 13 compounds described, respectively), while only four spiro-tetraflavonoids and two spiro-triflavonoids were described. They have been isolated from eight plant families represented by 46 species (Table 1). The main sources of spiro-flavonoids are plants belonging to Asparagaceae (34 compounds), Pinaceae (16) and Thymelaeaceae (10) families, especially species from Yucca (18 compounds), Larix (10), Abies (9), Drimiopsis (8) and Daphne (7) genera. In the case of Asparagaceae, most of the compounds (sixteen) belong to the group of scillascillin-type homoisoflavonoids, thirteen to spiro-flavostilbenoids and five to spiro-biflavonoids. On the other hand, Pinaceae and Thymelaeaceae were predominantly spiro-biflavonoid sources, as were Cupressaceae and Pentaphylacaceae. Both woody aerial parts and roots were used to isolate spiro-biflavonoids and spiro-flavostilbenoids. Spiro-tri and spiro-tetraflavonoids were isolated from twigs and bark, and the main sources of scillascillin-type homoisoflavonoids were bulbs.
Table 1. Spiro-flavonoids described between 1973 and 2022.
|
Family |
Plant |
Part |
Compound |
References |
|
Asparagaceae |
Bessera elegans Schult.f. |
bulbs |
57 |
[13] |
|
|
Chionodoxa luciliae Boiss. |
bulbs |
49-52 |
[14] |
|
|
Drimiopsis barteri Baker |
bulbs and leaves |
50, 56, 60, 61, 64 |
[15] |
|
|
Drimiopsis burkei Baker |
bulbs |
53, 62 |
[15] |
|
|
Drimiopsis maculata Lindl. & Paxton |
bulbs |
52, 56 |
[16,17] |
|
|
Eucomis schijffii Reyneke |
bulbs |
49 |
[17,18] |
|
|
Furcraea bedinghausii K.Koch |
roots |
38, 39 |
[19] |
|
|
Ledebouria graminifolia (Baker) Jessop |
bulbs |
58, 61 |
[20] |
|
|
Ledebouria hyderabadensis M.V.Ramana, Prasanna & Venu |
bulbs |
49 |
[21] |
|
|
Ledebouria ovatifolia (Baker) Jessop |
bulbs |
49, 50 |
[22] |
|
|
Ledebouria socialis (Baker) Jessop |
bulbs |
63, 64 |
[22] |
|
|
Merwilla natalensis (Planch.) Speta |
bulbs |
49 |
[23] |
|
|
Muscari armeniacum H.J.Veitch |
bulbs |
52, 54 |
[24] |
|
|
Muscari botryoides (L.) Mill. |
bulbs |
52, 54, 56 |
[24] |
|
|
Muscari comosum (L.) Mill. |
bulbs |
53 |
[25] |
|
|
Muscari neglectum Guss. ex Ten. |
bulbs |
53-56 |
[26] |
|
|
Scilla scilloides (Lindl.) Druce |
bulbs |
49, 51, 52, 59 |
[27] |
|
|
Yucca gloriosa L. |
roots |
38-48 |
[11,28,29] |
|
|
Yucca schidigera Roezl ex Ortgies |
bark |
1, 5-7, 29, 36-41, 44, 46-48 |
[30–33] |
|
Cistaceae |
Fumana procumbens (Dunal) Gren. & Godr. |
whole plant |
23 |
[34] |
|
Cupressaceae |
Glyptostrobus pensilis (D.Don) K.Koch |
trunk bark |
1-4, 10, 11 |
[35] |
|
Fabaceae |
Caesalpinia sappan L. |
heartwood |
65 |
[36] |
|
Pentaphylacaceae |
Anneslea fragrans Wall. |
twigs |
2, 8, 9, 30 |
[37,38] |
|
Pinaceae |
Abies chensiensis Tiegh. |
aerial parts |
1-3 |
[39] |
|
|
Abies delavayi Franch. var. delavayi |
aerial parts |
1 |
[40] |
|
|
Abies georgei Orr |
aerial parts |
1, 12 |
[41] |
|
|
Abies sachalinensis (F.Schmidt) Mast. |
bark |
1, 4, 12-17 |
[42] |
|
|
Larix decidua Mill. |
bark |
1-3 |
[43] |
|
|
Larix gmelinii (Rupr.) Kuzen. |
bark |
1, 14, 18, 31 |
[6,44,45] |
|
|
Larix olgensis Henry var. koreana Nakai |
bark |
14, 16, 19-21 |
[46] |
|
|
Larix sibirica Ledeb. |
bark |
1, 14, 31 |
[44] |
|
|
Pinus massoniana Lamb. |
bark |
35 |
[47] |
|
|
Tsuga longibracteata W.C.Cheng |
bark |
1, 2 |
[48] |
|
Thymelaeaceae |
Daphne aurantiaca Diels |
stem bark |
22, 24 |
[49] |
|
|
Daphne feddei H.Lév. |
stem bark |
22, 24, 26, 27 |
[50] |
|
|
Daphne genkwa Siebold & Zucc. |
roots |
23 |
[51] |
|
|
Daphne kiusiana Miq. |
stem |
22, 24 |
[52] |
|
|
Daphne kiusiana var. atrocaulis (Rehder) F.Maek. |
stem |
22 |
[53] |
|
|
Daphne linearifolia Hart |
aerial parts |
23-25, 28 |
[54] |
|
|
Daphne mucronata Royle |
shoots |
23 |
[55] |
|
|
Daphne odora Thunb. |
roots |
22, 24 |
[56,57] |
|
|
Edgeworthia chrysantha Lindl. |
stem and twigs |
22, 24, 32-34 |
[58] |
|
|
Stellera chamaejasme L. |
roots |
23 |
[59] |
|
|
Thymelaea microphylla Coss. & Durieu |
roots |
23 |
[60] |
|
|
Wikstroemia indica (L.) C.A.Mey. |
roots |
22, 23 |
[61,62] |
|
Vitaceae |
Vitis amurensis Rupr. |
seeds |
16 |
[63] |
Table 2. Chemical structures of spiro-biflavonoids.
|
|
|||||
|
Compound |
Name |
R1 |
R2 |
Stereochemistry |
|
|
C30H22O10 (MW = 542.49) |
|||||
|
1 |
Larixinol (= Abiesinol E) |
H |
H |
(2R,3R,2′R,3′R) |
|
|
2a |
3-Epi-larixinol |
H |
H |
(2R*,3R*,2′R*,3′S*) |
|
|
3a |
3,2′-Epi-larixinol |
H |
H |
(2S*,3R*,2′R*,3′S*) |
|
|
4 |
Abiesinol F |
H |
H |
(2R,3S,2′R,3′R) |
|
|
5 |
Yuccalechin A |
H |
H |
(2S,3R,2′R,3′R) |
|
|
6 |
Yuccalechin B |
H |
H |
(2S,3R,2′R,3′S) |
|
|
7 |
Yuccalechin C |
H |
H |
(2S,3S,2′R,3′S) |
|
|
8 |
Fragranol B |
H |
H |
(2S,3R,2′S,3′R) |
|
|
9 |
Fragranol C |
H |
H |
(2R,3S,2′S,3′R) |
|
|
10 |
Spiropensilisol A |
H |
H |
(2R*,3S*,2′R*,3′S*) |
|
|
11 |
Spiropensilisol B |
H |
H |
(2S*,3S*,2′R*,3′S*) |
|
|
C30H22O11 (MW = 558.49) |
|||||
|
12a |
Abiesinol A (= 13-Hydroxylarixinol) |
H |
OH |
(2R,3R,2′R,3′R) |
|
|
13 |
Abiesinol B |
H |
OH |
(2R,3S,2′R,3′R) |
|
|
14 |
Abiesinol C (= Olgensisinol A) |
OH |
H |
(2R,3R,2′R,3′R) |
|
|
15 |
Abiesinol D |
OH |
H |
(2R,3S,2′R,3′R) |
|
|
C30H22O12 (MW = 574.49) |
|||||
|
16 |
Abiesinol G (= Vitisinol) |
OH |
OH |
(2R,3R,2′R,3′R) |
|
|
17 |
Abiesinol H |
OH |
OH |
(2R,3S,2′R,3′R) |
|
|
18 |
Larisinol |
OH |
OH |
(2R*,3R*,2′R*,3′S*) |
|
|
19 |
Olgensisinol B |
OH |
OH |
(2R,3S,2′S,3′S) |
|
|
|
|||||
|
Compound |
Name |
Stereochemistry |
|||
|
C30H22O12 (MW = 574.49) |
|||||
|
20 |
Olgensisinol C |
(2R*,3S*,2′R*,3′S*) |
|||
|
21 |
Olgensisinol D |
(2R*,3R*,2′S*,3′R*) |
|||
|
|
|||||
|
Compound |
Name |
R1 |
R2 |
R3 |
Stereochemistry |
|
C30H22O9 (MW = 526.49) |
|||||
|
22 |
Daphnodorin C |
H |
H |
H |
(2S,3S,2′S) |
|
C30H22O10 (MW = 542.49) |
|||||
|
23 |
Genkwanol A |
OH |
H |
H |
(2R,3R,2′R,3′S) |
|
24 |
Daphnodorin I |
OH |
H |
H |
(2S,3S,2′R,3′S) |
|
C31H24O10 (MW = 556.52) |
|||||
|
25a |
4′-Methylgenkwanol A |
OH |
H |
CH3 |
(2R,3S,2′R,3′S) |
|
26a |
2′′-Methoxy-daphnodorin C |
H |
OCH3 |
H |
(2R*,3S*,2′R*) |
|
27a |
2′′-Methoxy-2-epi-daphnodorin C |
H |
OCH3 |
H |
(2R*,3S*,2′S*) |
|
C30H22O11 (MW = 558.49) |
|||||
|
28a |
2′′-Hydroxygenkwanol A |
|
|
|
(2R,3S,2′R,3′S) |
|
|
|||||
|
Compound |
Name |
Stereochemistry |
|||
|
C29H22O9 (MW = 514.48) |
|||||
|
29 |
Yuccaone A |
(2R*,4R*,2′S*,3′R*) |
|||
|
a The name of the compound refers to the name given in the original publication, while the names according to the numbering in the figure are: 3′-epi-larixinol (2), 3′,2-epi-larixinol (3), 3″-hydroxylarixinol (12), 4″-methylgenkwanol A (25), 2-methoxy-dpahnodorin C (26), 2-methoxy-2′-epi-daphnodorin C (27) and 2-hydroxygenkwanol (28). |
|||||
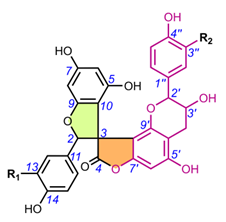
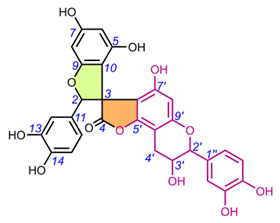
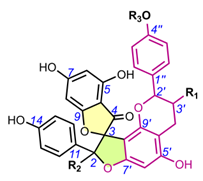
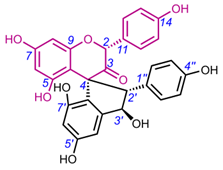
Table 3. Chemical structures of spiro-triflavonoids.
|
|
||
|
Compound |
Name |
Stereochemistry |
|
C45H30O15 (MW = 810.71) |
||
|
30 |
Fragranol A |
(2S,3S,2′S,3′R,2′′R,3′′S) |
|
|
||
|
31 |
Triflarixinol |
(2R*,3R*,2′R*,3′R*,2‴S*,3‴S*) |
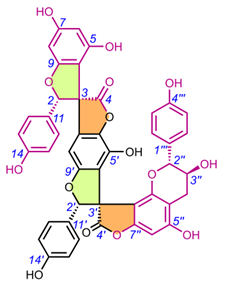
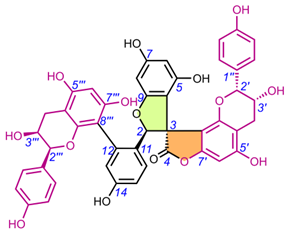
Table 4. Chemical structures of spiro-tetraflavonoids.
|
|
|||
|
Compound |
Name |
R |
Stereochemistry |
|
C60H42O18 (MW = 1050.97) |
|||
|
32 |
Edgechrin A |
H |
(2S*,3S*,2′S*,4′R*,2‴S*) |
|
C60H42O19 (MW = 1066.97) |
|||
|
34 |
Edgechrin B |
OH |
(2S*,3S*,2′S*,4′R*,2‴R*,3‴S*) |
|
|||
|
Compound |
Name |
|
Stereochemistry |
|
C60H42O18 (MW = 1050.97) |
|||
|
33 |
Edgechrin D |
|
(2S*,3S*,2′S*,2‴S*,4‴R*) |
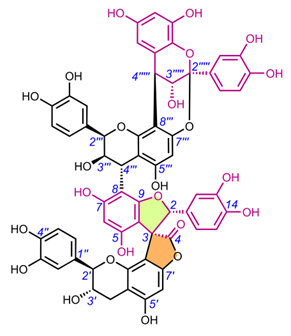
|
|||
|
Compound |
Name |
|
Stereochemistry |
|
C60H44O24 (MW = 1148.98) |
|||
|
35 |
Pinuspirotetrin |
|
(2R,3S,2′R,3′S,2‴R,4‴R,2‴″S,3‴″R,4‴″R) |
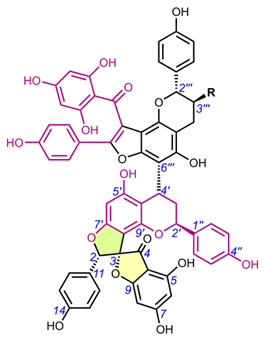
Table 5. Chemical structures of spiro-flavostilbenoids.
|
|
|||||
|
Compound |
Name |
R1 |
R2 |
R3 |
Stereochemistry |
|
C29H20O8 (MW = 496.47) |
|||||
|
36 |
Yuccaol A |
H |
OH |
H |
(2S,3R) |
|
37 |
Yuccaol B |
H |
OH |
H |
(2S,3S) |
|
C30H22O10 (MW = 542.49) |
|||||
|
38 |
Yuccaol C |
OH |
OCH3 |
H |
(2S,3S) |
|
39 |
Yuccaol D |
OH |
OCH3 |
H |
(2S,3R) |
|
40 |
Yuccaol E |
OH |
H |
OCH3 |
(2S,3S) |
|
41 |
Yuccalide A |
OH |
H |
OCH3 |
(2S,3R) |
|
|
|||||
|
Compound |
Name |
R1 |
R2 |
R3 |
Stereochemistry |
|
C30H22O10 (MW = 542.49) |
|||||
|
42 |
Yuccalide B |
OH |
OCH3 |
H |
(2R*,3R*) |
|
43 |
Yuccalide C |
OH |
OCH3 |
H |
(2R*,3S*) |
|
|
|||||
|
Compound |
Name |
Stereochemistry |
|||
|
C45H30O15 (MW = 810.71) |
|||||
|
44 |
Gloriosaol A |
(2S,3S,2′S,3′S) |
|||
|
45 |
Gloriosaol B |
(2S*,3S*,2′R*,3′R*) |
|||
|
46 |
Gloriosaol C |
(2S,3R,2′S,3′R) |
|||
|
47 |
Gloriosaol D |
(2S,3S,2′S,3′R) |
|||
|
48 |
Gloriosaol E |
(2S,3R,2′S,3′S) |
|||
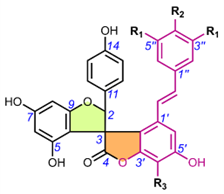
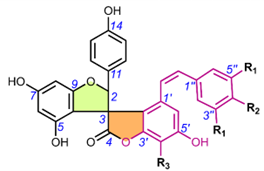
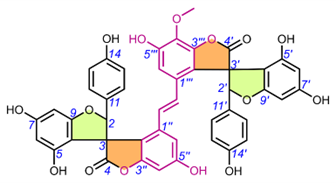
Table 6. Chemical structures of scillascillin-type homoisoflavonoids. Protosappanin D (65) is a dimeric protosappanin-type homoisoflavonoids.
|
|
||||||||
|
Compound |
Name |
R1 |
R2 |
Stereochemistry |
||||
|
C17H12O6 (MW = 312.27) |
||||||||
|
49 |
Scillascillin |
H |
H |
(3R) |
||||
|
C18H14O7 (MW = 342.30) |
||||||||
|
50 |
2-Hydroxy-7-O-methyl-scillascillin |
OH |
CH3 |
rac-(2R*,3R) |
||||
|
C17H12O7 (MW = 328.27) |
||||||||
|
51 |
2-Hydroxy-scillascillin |
OH |
H |
(3R*) |
||||
|
|
||||||||
|
Compound |
Name |
R1 |
R2 |
R3 |
R4 |
R5 |
R6 |
Stereochemistry |
|
C17H14O6 (MW = 314.29) |
||||||||
|
52 |
3′,5,7-Trihydroxy-4′-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (= isomuscomosin) |
H |
OH |
CH3 |
H |
H |
H |
(3R) |
|
53 |
4′,5,7-Trihydroxy-3′-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (= muscomosin) |
H |
OCH3 |
H |
H |
H |
H |
(3R) |
|
54 |
3′,4′,5-Trihydroxy-7-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
H |
OH |
H |
H |
CH3 |
H |
(3R) |
|
C18H16O6 (MW = 328.32) |
||||||||
|
55 |
5,5′-Dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
H |
H |
CH3 |
OH |
CH3 |
H |
(3R) |
|
56 |
3′,5-Dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
H |
OH |
CH3 |
H |
CH3 |
H |
(3R) |
|
57 |
5,5′,7-Trihydroxy-4′-methoxy-6-methylspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]-trien]-4-one |
H |
H |
CH3 |
OH |
H |
CH3 |
(3R) |
|
58 |
5,7-Dihydroxy-3′,4′-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
H |
OCH3 |
CH3 |
H |
H |
H |
(3R*) |
|
C18H16O7 (MW = 344.32) |
||||||||
|
59 |
2′,5,7-Trihydroxy-3′,4′-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (= scillavone A) |
OH |
OCH3 |
CH3 |
H |
H |
H |
(3R) |
|
60 |
2′,4′,5-Trihydroxy-3′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
OH |
OCH3 |
H |
H |
CH3 |
H |
(3R*) |
|
C19H18O6 (MW = 342.34) |
||||||||
|
61 |
5-Hydroxy-3′,4′,7-trimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
H |
OCH3 |
CH3 |
H |
CH3 |
H |
(3R*) |
|
C19H18O7 (MW = 358.34) |
||||||||
|
62 |
5,7-Dihydroxy-2′,3′,4′-trimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
OCH3 |
OCH3 |
CH3 |
H |
H |
H |
(3R*) |
|
63 |
2′,5-Dihydroxy-3′,4′,7-trimethoxyspiro[4H-1-benzopyran-3(2H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (= socialinone) |
OH |
OCH3 |
CH3 |
H |
CH3 |
H |
(3R*) |
|
C20H20O7 (MW = 372.37) |
||||||||
|
64 |
5-Hydroxy-2′,3′,4′,7-tetramethoxyspiro[4H-1-benzopyran-3(2H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one |
OCH3 |
OCH3 |
CH3 |
H |
CH3 |
H |
(3R*) |
|
|
||||||||
|
Compound |
Name |
Stereochemistry |
||||||
|
C32H28O12 (MW = 604.56) |
||||||||
|
65 |
Protosappanin D |
Unknown |
||||||
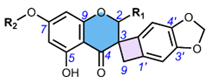
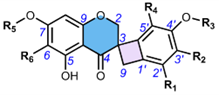
3. Methods of Extraction and Isolation
Due to their differing lipophilicity, spiro-flavonoids were extracted using different organic solvents and their mixtures, mainly with water (Table 7). Therefore, spiro-biflavonoids were extracted most frequently with EtOH, MeOH and their aqueous solutions (in more than 2/3 of the publications); spiro-triflavonoids with ethyl acetate (EtOAc) or an aqueous solution of EtOH; spiro-tetraflavonoids with an aqueous solution of acetone; and spiro-flavostilbenoids with MeOH and its aqueous solutions or acetone. On the other hand, scillascillin-type homoisoflavonoid were extracted from plant material using dichloromethane (DCM), its solution with MeOH, or MeOH alone, but also diethyl ether. In general, MeOH, EtOH, and their aqueous solutions were the extractants most commonly used. The extraction process was usually carried out by maceration at room temperature (28 papers), 15 articles reported extraction at boiling point (heat-reflux method), and the remaining 13 publications did not provide this information. Column chromatography (CC) and liquid–liquid extraction technique (LLE) were frequently used as the first step of the compound isolation procedure, and the LLE-fraction obtained with EtOAc was used in further steps. The pre-purification of crude extracts by CC was most commonly performed by normal phase, followed by reversed phase, and much less frequently by gel filtration chromatography (GPC). Normal-phase CC mainly used gradients of many different solvents: CHCl3, DCM, diethyl ether, EtOAc, n-hexane, and MeOH. Reversed-phase CC almost exclusively used gradients of MeOH and H2O, while MeOH was used for CC GPC . Interestingly, in the first stage of spiro-biflavonoid isolation, LLE was used more extensively than CC, while the opposite was true for scillascillin-type homoisoflavonoids. The next isolation step involved the use of CC (mainly normal-phase), regardless of the spiro-compound group, followed by the use of thin layer chromatography (TLC), medium pressure, and high performance liquid chromatography (MPLC and HPLC). The final isolation steps usually involved CC combined with MPLC, HPLC, and TLC.
Table 7. Summary of spiro-flavonoid isolation methods.
|
Group |
Compound |
Extraction solvent |
Isolation 1st step |
Isolation 2nd step |
Isolation final steps |
References |
|
Spiro-biflavonoids |
1 |
Acetone (maceration) |
LLE H2O-CHCl3-EtOAc* |
CC GPC LH-20 (EtOH) |
CC NP Silica gel (CHCl3-MeOH) |
[45] |
|
22, 24 |
Acetone-H2O (7:3, v/v) (maceration, r.t.) |
CC RP HP20 (gr. MeOH-H2O) |
CC GPC LH-20 (gr. MeOH-H2O) for 22 or CC RP MCI (gr. MeOH-H2O) for 24 |
CC RP MCI (MeOH-H2O) → CC RP C8 (MeOH-H2O) for 22 or CC RP HW-40F (MeOH-H2O) for 24 |
[58] |
|
|
|
1, 4, 12-17 |
CHCl3 → MeOH (maceration, r.t., successively) |
CC RP Diaion HP-20 (MeOH-H2O) |
CC NP Silica gel (gr. CHCl3-MeOH) |
MPLC NP Silica gel → CC GPC LH-20 (MeOH) → HPLC RP C18 (MeOH-H2O) |
[42] |
|
|
23 |
CHCl3-MeOH (2:1, v/v) (reflux) |
CC RP C18 (gr. MeOH-H2O) |
FLASH CC NP Silica gel (gr. CHCl3-MeOH-Acetone) |
FLASH CC NP Silica gel (Hexanes-EtOAc-MeOH) → CC NP Silica gel (EtOAc-CHCl3-MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) |
[55] |
|
|
1, 14, 18, 22, 24 |
EtOAc (reflux) |
CC NP Silica gel (gr. CHCl3-MeOH) for 1, 14, 18 or CC NP Silica gel (gr. n-Hexane-EtOAc) for 22, 24 |
CC NP Silica gel (CHCl3-MeOH) or CC GPC LH-20 (MeOH) for 22 |
CC GPC LH-20 (MeOH) for 24 |
[6,44,56,57] |
|
|
23 |
EtOH (r.t.) |
LLE H2O-Petroleum ether-CHCl3-EtOAc |
CC NP Silica gel (gr. CHCl3-MeOH) |
|
[59] |
|
|
1-3 |
EtOH-H2O (40%, v/v) (stirring, r.t.) |
HPLC RP C18 (CH3CN-H2O w/ FA) |
|
|
[43] |
|
|
23 |
EtOH-H2O (7:3, v/v) (maceration, r.t.) |
LLE H2O-Petroleum ether-DCM-EtOAc-n-BuOH |
CC NP Silica gel (gr. Petroleum Ether-EtOAc-MeOH) |
CC NP Silica gel (gr. DCM-MeOH) → CC GPC LH-20 (MeOH) → TLC RP (MeOH-H2O) |
[60] |
|
|
23 |
EtOH-H2O (75%, v/v) (reflux) |
LLE H2O-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-MeOH) |
HPLC RP C18 (MeOH-H2O) |
[61] |
|
|
1-3 |
EtOH-H2O (80%, v/v) |
LLE H2O-CHCl3-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-MeOH) |
CC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) |
[39] |
|
|
14, 16, 19-21 |
EtOH-H2O (80%, v/v) |
LLE H2O-CHCl3-EtOAc |
CC RP XAD-7 HP (gr. MeOH-H2O) |
CC GPC LH-20 (gr. MeOH-H2O) → CC RP MCI gel CHP-20P (EtOH-H2O) for 14, 16, 19 or CC GPC LH-20 (gr. MeOH-H2O) → CC GPC LH-20 (MeOH-H2O) → CC RP C18 (MeOH-H2O) for 20, 21 |
[46] |
|
|
16 |
EtOH-H2O (80%, v/v) |
LLE H2O-CHCl3-EtOAc-n-BuOH |
CC RP Diaion HP-20 (MeOH-H2O) |
CC GPC LH-20 (gr. MeOH-H2O) → CC RP MCI CHP-20P (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O w/ AcOH) |
[63] |
|
|
1, 12 |
EtOH-H2O (80%, v/v) (reflux) |
LLE H2O-CHCl3-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-Acetone) |
MPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) |
[41] |
|
|
1 |
EtOH-H2O (85:15, v/v) (maceration, r.t.) |
LLE H2O-CHCl3-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-Acetone) |
MPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) |
[40] |
|
|
2, 8, 9 |
EtOH-H2O (95%, v/v) (maceration, r.t.) |
LLE H2O-EtOAc |
MPLC NP Silica gel (DCM-EtOAc) |
TLC NP Silica gel (DCM-EtOAc) |
[38] |
|
|
22 |
EtOH-H2O (95%, v/v) (reflux) |
LLE H2O-Petroleum ether-EtOAc |
CC NP Silica gel (gr. DCM-MeOH) |
CC NP Silica gel (unknown solvent) → HPLC RP C18 (MeOH-H2O) |
[53] |
|
|
1, 2, 22 |
EtOH-H2O (95%, v/v) (r.t.) |
LLE H2O-Petroleum ether-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-MeOH) for 22 or CC NP Silica gel (Petroleum Ether-Acetone) for 1, 2 |
CC GPC LH-20 (CHCl3-MeOH) for 22 or HPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) for 1, 2 |
[48,62] |
|
|
22, 24, 26, 27 |
MeOH |
LLE H2O-Petroleum ether-EtOAc-n-BuOH |
CC NP Silica gel (gr. CHCl3-MeOH) |
CC RP C18 (gr. MeOH-H2O) → CC GPC LH-20 (MeOH) or CC GPC LH-20 (MeOH) for 22, 24 |
[49,50] |
|
|
22, 24 |
MeOH(maceration, r.t.) |
LLE H2O-n-Hexane-EtOAc-n-BuOH |
CC RP C18 (MeOH-H2O) |
HPLC RP C18 (CH3CN-H2O) |
[52] |
|
|
29 |
MeOH (maceration, r.t.) |
CC GPC LH-20 (MeOH) |
CC RP C18 (CH3CN-H2O w/ H3PO4) |
|
[32] |
|
|
23 |
MeOH (reflux) |
CC NP Silica gel(gr. n-Hexane-EtOAc → CHCl3-MeOH) |
CC GPC LH-20 (MeOH-H2O) |
CC RP MCI CHP 20P (MeOH-H2O) |
[51] |
|
|
1 |
MeOH (r.t.) |
CC RP C18 (gr. MeOH-H2O) |
CC RP C18 (gr. CH3CN-H2O) |
|
[33] |
|
|
5-7 |
MeOH (r.t.) |
LLE H2O-n-Hexane- EtOAc |
CC GPC LH-20 (MeOH) |
CC NP Silica gel (gr. CHCl3-Acetone-AcOH) → HPLC RP C18 (CH3CN/MeOH-H2O) |
[31] |
|
|
23 |
MeOH-H2O (80%, v/v) (37°C) |
LLE H2O-n-BuOH |
CC GPC LH-20 (MeOH) |
CC NP Silica gel (gr. cyclohexane-EtOAc-MeOH) → CC MPLC RP (CH3CN-H2O) |
[34] |
|
|
1-4, 10, 11 |
MeOH-H2O (90%, v/v) (r.t.) |
CC GPC LH-20 |
HPLC RP C18 |
|
[35] |
|
|
23-25, 28 |
n-Hexane → CHCl3 → CHCl3-MeOH (9:1, v/v) → MeOH (maceration, r.t., successively) |
CC GPC LH-20 (MeOH) |
HPLC RP C18 (MeOH-H2O) |
|
[54] |
|
Spiro-triflavonoids
|
31 |
EtOAc (reflux) |
CC NP Silica gel (gr. CHCl3-MeOH) |
CC NP Silica gel (CHCl3-MeOH) |
|
[44] |
|
30 |
EtOH-H2O (95%, v/v) (maceration, r.t.) |
LLE H2O-EtOAc |
MPLC NP Silica gel (gr. DCM-EtOAc-MeOH) |
MPLC NP Silica gel (gr. DCM-MeOH) |
[37] |
|
|
Spiro-tetraflavonoids |
32-34 |
Acetone-H2O (7:3, v/v) (maceration, r.t.) |
CC RP HP20 (gr. MeOH-H2O) |
CC GPC LH-20 (gr. MeOH-H2O) |
CC NP Silica gel (DCM-MeOH) → CC RP HW-40F (MeOH-H2O) → CC RP C8 (MeOH-H2O) → CC RP C18 (MeOH-H2O) for 32 or CC RP HW-40F (MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) for 33 or CC NP Silica gel (DCM-MeOH) → CC RP C18 (MeOH-H2O) for 34 |
[58] |
|
|
35 |
*Pine bark extract |
LLE H2O-EtOAc |
CPC Hexane-EtOAc-MeOH-H2O (2-4-1-4) → CPC Hexane-EtOAc-MeOH-H2O (0.5-4-1-4) |
CC GPC LH-20 (EtOH) → CC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O w/ FA) |
[47] |
|
Spiro-flavostilbenoids |
38-43 |
Acetone (r.t.) |
CC RP DMS (gr. MeOH-H2O-Acetone) |
CC GPC LH-20 (MeOH) |
HPLC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) |
[11] |
|
38, 39 |
MeOH |
LLE H2O-EtOAc-n-BuOH |
CC NP Silica gel (gr. n-Hexane-EtOAc-MeOH) |
CC NP Silica gel (n-Hexane-EtOAc) → CC GPC LH-20 (MeOH) |
[19] |
|
|
|
36-40 |
MeOH (r.t.) |
CC RP C18 (gr. MeOH-H2O) |
CC RP C18 (gr. CH3CN-H2O H3PO4) or CC RP C18 (gr. CH3CN-H2O) |
|
[30,33] |
|
|
41, 44, 46-48 |
MeOH (r.t.) |
LLE H2O-n-Hexane-EtOAc |
CC GPC LH-20 (MeOH) |
CC NP Silica gel (gr. CHCl3-Acetone-AcOH) → HPLC RP C18 (CH3CN/MeOH-H2O) |
[31] |
|
|
38-40, 44-48 |
MeOH-H2O (80%, v/v) |
LLE H2O-EtOAc |
CC GPC LH-20 (MeOH) |
HPLC RP C18 (gr CH3CN-H2O w/ TFA) |
[28,29] |
|
Scillascillin-type homoisoflavonoids |
49, 52, 56 |
DCM (agitation or shaker, r.t.) |
CC NP Silica gel (unknown solvent or DCM) |
|
|
[16,23] |
|
49, 50 |
DCM → EtOAc → MeOH (shaker, successively) |
CC NP Silica gel (n-Hexane-DCM-MeOH) |
CC NP Silica gel (DCM-Et2O) |
|
[22] |
|
|
|
49 |
DCM → MeOH (agitation, r.t.) |
CC NP Silica gel (DCM) |
|
|
[18] |
|
|
63, 64 |
DCM → MeOH (shaker, successively) |
CC NP Silica gel (gr. n-Hexane-DCM-MeOH) |
TLC NP Silica gel (EtOAc-DCM) |
|
[22] |
|
|
50, 56, 60-62, 64 |
DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) |
LLE MeOH-CHCl3-H2O (lower phase) |
CC NP Silica gel (gr. DCM-MeOH) |
CC NP Silica gel (gr. CHCl3-Light Petroleum) → recrystallization (MeOH) for 50 or CC GPC LH-20 (unknown solvent) → CC NP Silica gel (gr. CHCl3-MeOH) → TLC (unknown solvent) for 60 or CC GPC LH-20 (unknown solvent) → CC NP Silica gel (gr. CHCl3-Light Petroleum) → TLC (CHCl3-Light Petroleum-MeOH) for 56, 61, 62, 64 |
[15] |
|
|
53 |
DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) |
LLE (MeOH-H2O, 70%, v/v)-Light Petroleum-CHCl3-EtOAc-n-BuOH (lower phase) |
CC NP Silica gel (gr. Light Petroleum-EtOAc) |
CC NP Silica gel (gr. Light Petroleum-CHCl3-MeOH) |
[15] |
|
|
58, 61 |
DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) |
CC NP Silica gel (gr. Petrol-EtOAc-MeOH) |
TLC NP Silica gel (CHCl3-MeOH) |
TLC NP Silica gel (CHCl3) |
[20] |
|
|
52, 54, 56 |
Light petrol → Et2O → MeOH (Soxhlet, successively) |
CC NP Silica gel (gr. n-Hexane-Et2O-MeOH) |
CC NP Silica gel (gr. n-Hexane-Acetone) |
HPLC NP Silica gel (CHCl3-MeOH) → PLC NP Silica gel (CHCl3-Acetone) |
[24] |
|
|
53-56 |
Light petrol → Et2O → MeOH (Soxhlet, successively) |
CC NP Silica gel (CHCl3-EtOAc) |
PLC NP Silica gel (Benzene-EtOAc) → PLC NP Silica gel (CHCl3-Acetone) or PLC NP Silica gel (Benzene-EtOAc) → crystallization (MeOH) |
|
[26] |
|
|
57 |
MeOH (reflux) |
CC RP Diaion HP20 (gr. MeOH-H2O → MeOH → EtOH → EtOAc) |
CC NP Silica gel (gr CHCl3-MeOH-H2O) |
CC NP Silica gel (CHCl3-MeOH-H2O) → CC RP C18 (MeOH-H2O) |
[13] |
|
|
49, 51, 52, 59 |
MeOH (r.t.) |
LLE H2O-EtOAc |
CC NP Silica gel (gr. n-Hexane-Acetone) |
CC RP C18 (gr. MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → crystallization (n-Hexane-Acetone) or CC RP C18 (gr. MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → HPLC NP Silica gel (CHCl3-MeOH) |
[27] |
|
|
53 |
Petrol → Et2O (Soxhlet, successively) |
CC NP Silica gel (gr. n-Hexane-Et2O) |
CC NP Silica gel (gr. CHCl3-EtOAc) |
CC NP Silica gel (Benzene-EtOAc) → TLC NP Silica gel (n-Hexane-Et2O-dioxane) |
[25] |
|
|
49-52 |
Petrol → Et2O → MeOH (Soxhlet, successively) |
CC NP Silica gel (gr. CHCl3-MeOH) |
TLC NP Silica gel (Benzene-EtOAc) |
PLC NP Silica gel (Benzene-EtOAc) |
[14] |
|
* The fraction obtained with the solvent marked in bold and underlined font was used in further isolation steps; r.t. = room temperature; ** extraction solvent not disclosed; DCM = CH2Cl2; LLE = liquid-liquid extraction; CC = column chromatography; TLC = thin-layer chromatography; PLC = preparative thin-layer chromatography; HPLC = high-performance liquid chromatography; MPLC = medium pressure liquid chromatography; GPC = gel permeation chromatography; CPC = centrifugal partition chromatography; NP = normal-phase; RP = reversed-phase; FA = formic acid; TFA = trifluoroacetic acid. |
||||||
4. Stereochemistry of the Isolated Spiro-Flavonoids
The presence of up to 8 stereogenic centers in the known spiro-flavonoid molecules and the associated possibility of up to 128 pairs of enantiomers (28 = 256) occurring in nature makes it a challenging task to assign the relative configuration (RelC), let alone the absolute configuration (AbsC). When molecules with multiple stereogenic centers are studied, the determination of the relative configuration is particularly important for the assignment of the AbsC. In such a case, the determination of the AbsC by a single chiroptical spectroscopic method may lead to an incorrect assignment of the absolute configuration, especially if no prediction is made by quantum mechanical (QM) calculation methods and only by comparison with the spectra of previously described compounds (empirical methods). This is because in some cases, even QM (e.g., ECD)-calculated spectra may match more than one diastereoisomer, and interactions of different chromophores should be taken into account for correct AbsC assignment [64,65].
Spiro-flavonoids are compounds that contain 4-, 5-, and 6-membered rings in their structure. To determine the relative configuration in cyclic compounds with three- to six-membered rings, which exhibit a predictable conformational behavior, it is usually sufficient to extract some NMR parameters such as the 1H-1H vicinal coupling constants (3JHH) (and the corresponding dihedral angles via the Karplus equation [66]) and nuclear Overhauser effect spectroscopy (NOESY) correlations. Additional information can be obtained in systems containing electronegative substituents directly attached to the α-carbon (i.e., oxygen, nitrogen, or halogen) can be obtained by measuring the values of the heteronuclear coupling constants – if the value of 2JHC is large (4-7 Hz), the position of the electronegative substituent is gauche relative to the geminal proton, and becomes small (0-2 Hz) if it is antiperiplanar. On the other hand, 3JHC, similar to 3JHH, follows the Karplus relation and can also be used to determine additional constraints on dihedral angles – the values of this constant are lower (1-3 Hz) for gauche than for anti-periplanar conformation (5-8 Hz) [31,67–69]. An empirical rule developed by Nakashima et al. (2016) [11], which allows easy determination of the relative configuration for the spiro-carbon (C-3) and its neighboring carbon (C-2), was applied on the basis of the observation of 13C NMR chemical shifts for some spiro-flavonoids (abiesinols A-H, yuccalides A-C, yuccaols A-E, and gloriosaols A-E) reported in this review. Namely, for the 13C NMR shifts measured in the acetone-d6 and methanol-d4, the γ-lactone carbonyl group (C-4) located in the δC range of 174.6-176.9 indicates a syn-relationship between C-4 and the aromatic substituent located at the C-2 carbon, due to the anisotropic shielding effect exerted by the aromatic substituent on C-4, and thus the rel-(2R,3S) configuration. On the other hand, the γ-lactone carbonyl group, which has an anti-relationship with the aromatic substituent in C-2, has chemical shifts in the range δC 178.6-181.1, indicating a rel-(2R,3R) configuration. An additional tool, increasingly used for RelC determination, is the use of quantum-based Gauge-Independent Atomic Orbital (GIAO) NMR shift calculations via a modified DP4+ probability method, which allows one to determine with high probability which of the possible stereoisomers is the correct one [70].
The techniques used to determine AbsC are direct methods such as electronic and vibrational circular dichroism (ECD and VCD), but also indirect (or relative) methods such as circular dichroism using empirical methods, NMR with chiral derivatization agents [71] (e.g., Mosher's method [72,73]). The crux of Mosher's method lies in the difference in anisotropic effects between two diastereomers prepared from the substrate (molecule) of interest with a pair of enantiomeric chiral derivatizing agents (CDAs). In practice, the differences in chemical shifts (ΔδRS or ΔδSR) between the two substrate derivatives are used to assign AbsC. The most common CDA is MTPA (α-methoxy-α-trifluoromethylphenylacetic acid), commonly known as Mosher's reagent. This method typically involves the double derivatization of substrates with two CDA enantiomers and therefore requires a relatively large sample amount. In recent years, however, ECD has become a particularly popular method for determining AbsC. ECD involves an electron transition and can therefore be predicted theoretically by QM calculations. With the measured CD spectrum with the TDDFT (Time-Dependent Density-Functional Theory)-calculated CD spectrum of an assumed configuration, one can easily assign the AbsC of a chiral compound. Another advantage of this technique is that only sub-μg amounts of the sample needed to obtain the spectrum, which measures the difference in the chiral response of a molecule to UV/Vis modulation between the left and right polarized states [74]. To our knowledge, X-ray diffraction (XRD), which is a direct method that can be used to determine AbsC and is considered to be very reliable [75], has only been used for the determination of RelC of spiro-flavonoids.
4.1. Spiro-Biflavonoids
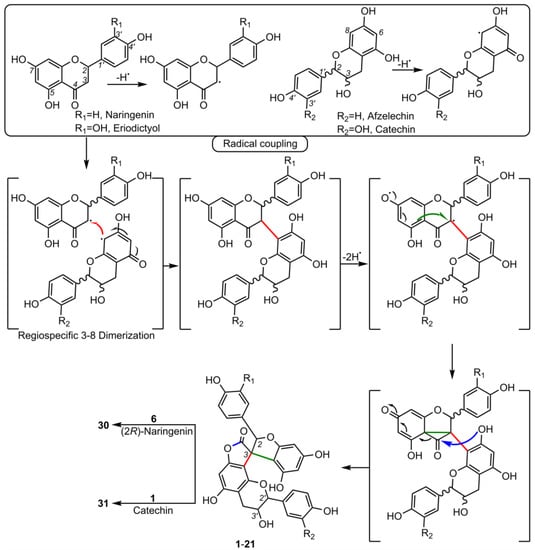
4.1.1. Larixinol Sub-Group (1-21) and Yuccaone A (29)
One of the first spiro-biflavonoids discovered was larixinol (1) isolated from the bark of Larix gmellini (Rupr.) Kuzen. [45,76]. Its structure (Table 2) and its relative configuration were assigned by X-ray crystallographic analysis as rel-(2R,3R,2′R,3′R), but its absolute configuration was not confirmed until almost a quarter of a century later by Wada et al. (2009), who isolated abiesinol E (= larixinol) from the bark of Abies sachalinensis (F.Schmidt) Mast., and assigned its absolute configuration as 2R,3R,2′R,3′R using Mosher’s method [42]. A total of eight abiesinols (A-H) (1, 4, 12-17) were reported in the same article. Their relative configurations were determined using 3JHH coupling constants and NOEs, and their AbsCs were determined using the Mosher method and by comparing optical rotations and ECD spectral data. Interestingly, abiesinol G (16) had the same chemical structure and stereochemistry as vitisinol first isolated from the seeds of Vitis amurensis Rupr. [63], while abiesinol C = olgiensisinol A (14) first isolated from stem bark of Larix olgensis Henry var. koreana Nakai [46] (Table 2). In the latter work, the authors used spatial anisotropic shielding effects for the γ-lactone carbon and NOESY to determine the RelC, and a comparison of ECD spectra with the literature data was used to determine the AbsC of olgensisinol A and B (19). The authors of this work also reported the relative configuration of other unusual spiro-biflavonoids, olgiensisinols C and D (20, 21), but no attempt was made to determine their AbsC. Yuccalechins A-C (5-7) (Table 2), extracted from the bark of Yucca schidigera and described by Pecio et al. (2019) [31], are another example of compounds whose AbsC was determined with high reliability using measurements of ECD spectra and TDDFT of calculated ECD spectra. In this publication, the authors used measurements of different coupling constants (3JHH, 1JHC, 2JHC, and 3JHC), spatial anisotropic shielding effects for 13C NMR chemical shifts, and NOESY correlations for the determination of RelC as well as QM calculations with the DP4+ method. Similarly, spatial anisotropic shielding effects of 13C NMR and NOESY correlations were used to determine RelC, and QM calculations of ECD spectra were used to determine AbsC of fragranols B and C (8, 9) (Table 2) by Omar et al. (2020) [38].
Several papers have also been published in which RelC / AbsC have been determined in a way that casts doubt on their accuracy. For example, Li et al. (2009) [39] report 3-epi-larixinol (2) and 3,2′-epi-larixinol (3) whose AbsC were implicitly suggested using 3JHH coupling constants, NOESY correlations and optical rotations. Unfortunately, the authors did not measure ECD spectra and nor did they use Mosher’s method, which could be critical to determining the stereochemistry. Following this line of thought, it turns out that if the absolute configuration of 3,2′-epi-larixinol, i.e. (2S,3R,2R,3S), is taken as true, it corresponds exactly to yuccalechin B (6), but its optical rotations differ in sign and value (+25.0 vs. -175.0, respectively), and the chemical shifts of 1H and 13C differ significantly, e.g., for H-2 / C-2, C-4 and H-2 / C-2' (δH / δC 5.79 / 95.3, 181.0, 4.57 / 83.1 vs. 6.13 / 90.6, 177.1, 4.99 / 81.6). Another example that raises questions about the use of empirical methods to determine AbsC is the work of Xiong et al. (2020) [35], which describes spiropensilisol A and B (10, 11). The relative configurations of these compounds were determined using 3JHH coupling constants, 13C NMR anisotropic shielding effects, and NOEs. Using this method, the authors proposed the AbsC of spiropensilisol B as (2S,3S,2'R,3'S)-11, which coincides with the configuration of yuccalechin C (7). However, the ECD spectra show a significant difference around 220 nm between these two compounds. On the other hand, based only on 1H and 13C NMR chemical shifts and optical rotations, Yang et al. (2011) [41] proposed 13-hydroxylarixinol as a new natural compound isolated from Abies georgei Orr. Assuming that the AbsC determination for this compound by this route is correct, its absolute configuration corresponds to that of abiesinol A (12), which questions the novelty of this discovery. Similarly, in the work of Piacente et al. (2004) [33], the authors reported the presence of larixinol only on the basis of 1H and 13C NMR values, while Pecio et al. (2019) [31] did not confirm the presence of larixinol, but reported three compounds in the same plant with the same planar structure but different stereochemistry, i.e., yuccalechins A-C. The presence of an unusual spiro-biflavonoid, yuccaone A (29), was also reported in the bark of Y. schidigera [32,33], the RelC of which was determined by NOESY correlation as (2R*,4R*,2'S*,3'R*). On the other hand, Fedorova et al. (2007) [6] only partially assigned the RelC of spiro-biflavonoid larisinol (18) using 1H/13C NMR analysis.
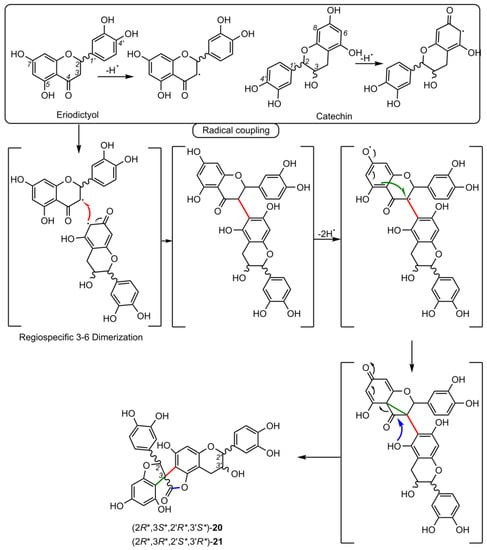
4.1.2. Daphnodorin C Sub-Group (22-28).
Daphnodorin C (22) is the simplest representative of a group of compounds isolated from a Daphne Tourn. ex L. genus, with a chemical structure quite similar to the larixinol group (Table 2). However, instead of a γ-butyrolactone ring, it has a 3-oxotetrahydrofuran ring, indicating a different biosynthetic pathway. This compound, first described by Baba et al. in 1986, was isolated from the roots and bark of Daphne odora Thunb. [77]. Its relative configuration was determined by XRD and NOESY correlations, and AbsC (2S,3S,2′S) was assigned by chemical degradation (H2SO4/EtOH), resulting in a product with a known absolute configuration (daphnodorin A) [78]. Another representative of this group of compounds is genkwanol A (23), isolated from the roots of Daphne genkwa Siebold & Zucc. [51]. Its structure and AbsC (2R,3R,2′R,3′S) were determined by Baba et al., 1987, by chemical transformation (HCl/MeOH) to daphnodorin B and comparison of Cotton effects observed in the ECD spectra [78]. AbsC (2S,3S,2′R,3′S) of daphnodorin I (24), which is a diastereoisomer of genkwanol A, was determined in a similar manner, i.e., by chemical conversion of 24 to daphnodorin B (using HCl/MeOH), and comparison of their ECD spectra [57]. Malafronte et al. (2012) isolated and elucidated the structures of 4′-methylgenkwanol A (25) and 2″-hydroxygenkwanol A (28) from Daphne linearifolia Hart. The compounds had identical AbsC, (2R,3S,2′R,3′S), determined by comparison of 1H/13C and ECD data with those of genkwanol A [54]. 2″-Methoxy-daphnodorin C (26) and 2″-methoxy-2-epi-daphnodorin C (27) were isolated from Daphne feddei H.Lév. and their stereochemistry was determined by NMR data consistent with daphnodorin C and XRD analysis, which confirmed their RelC as (2R*,3S*,2′R*)-26 and (2R*,3S*,2′S*)-27, respectively [50].
4.2. Spiro-Triflavonoids (30-31)
Fragranol A, a spiro-triflavonoid containing two spiro-centers and six chiral carbon atoms, was isolated from twigs of Anneslea fragrans (Table 3) [37]. The RelC of the comp. 30 was determined by NOESY correlations and spatial anisotropic shielding effects around the spiro-centers. Fragranol A AbsC was confirmed by QM ECD calculations and assigned as (2S,3S,2′S,3′R,2″R,3″S). Ivanova et al. (2006) partially assigned the RelC of spiro-triflavonoid triflarixinol (31) using 1H/13C NMR analysis, infrared spectroscopy, and melting point measurements of recrystallized compounds [44]. However, the authors did not attempt to determine the AbsC.
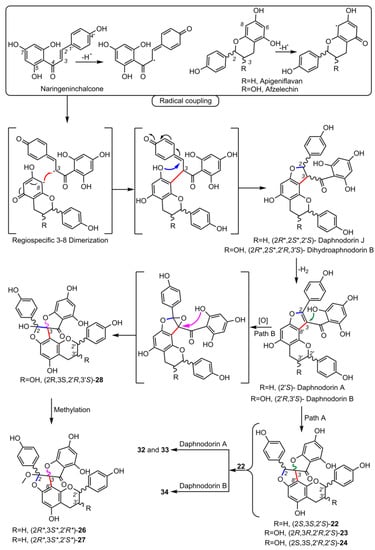
4.3. Spiro-Tetraflavonoids (32-35)
Edgechrins (Table 4) are condensed daphnodorin dimers – daphnodorin C-(4′β→6‴)-daphnodorin A, daphnodorin C-(4′β→6‴)-daphnodorin B, daphnodorin A-(4‴β→8)-daphnodorin C (comp. 32-34, respectively), and their structures were elucidated by the 1H-, 13C- and DEPT, COSY and NOESY NMR spectra [58]. The HMBC correlations were used to determine the linkage between dimers. Their AbsC were assigned by biogenetic considerations and supported by the empirical method: comparison of Cotton effects observed in the ECD spectra. The ECD spectra were compared with daphnodorins A, B, and C. The authors report that they performed NOESY NMR measurements. However, they do not present the use of NOE to determine RelC or to confirm AbsC against known biogenetic monomers. Furthermore, the absolute configurations of the structures are not explicitly reported. Moreover, the absolute configurations in the text of the article did not match the stereochemistry of the structures shown in the figures, which seems to undermine the correctness of the stereochemistry determination. Zhou et al. (2020) [47] reported another interesting tetrameric spiro-flavonoid, pinuspirotetrin (35), which they described as a proanthocyanidin. It was isolated from the bark of Pinus massoniana Lamb. This compound represents the first heterodimeric (4→8 linked) proanthocyanidin containing a spiro-type and an A-type dimer. The authors determined the AbsC of this unusual structure in a plausible manner, using combined spectroscopic methods, i.e. NOESY NMR correlations and an empirical method based on the use of ECD spectra and Cotton effects for known compounds, to establish the stereochemistry of 35.
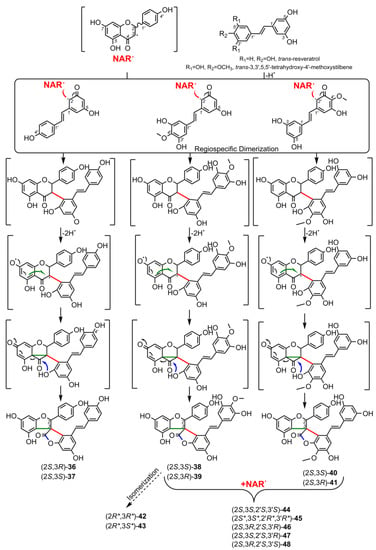
4.4. Spiro-Flavostilbenoids (36-48)
Spiro-flavostilbenoids are derivatives of trans-resveratrol (trans-3,4′,5-trihydroxystilbene) or trans-3,3′,5,5′-tetrahydroxy-4′-methoxystilbene (THMS) and naringenin. Structures of yuccaols A-E (36-40) were first described by Oleszek et al. (2001) [30] and Piacente et al. (2004) [33], while yuccalides A-C (41-43) and gloriosaols A-E (44-48) (Table 5) were described throughout the next decade [11,28,29]. The authors of these articles reported only the relative configurations for the isolated compounds – rel-(2R,3S) for compounds 36, 39, 41, 43, rel-(2R,3R) for compounds 37, 38, 40, 42, rel-(2R,3R,2′R,3′R) for 44, rel-(2S,3S,2′R,3′R) for 45, rel-(2R,3S,2′R,3′S) for 46, rel-(2R,3R,2′R,3′S) for 47, and rel-(2R,3S,2′R,3′R) for 48. These were determined by NOESY correlations and, in the case of gloriosols A-E, by QM geometry calculations and GIAO 1H chemical shifts. Chiroptical properties in the form of optical rotations have been measured for all members of the group, except for gloriosaols D and E isolated as a mixture. However, ECD spectra have been obtained for yuccalides A-C and yuccaols C-E. More recently, Pecio et al. (2023) [7] established AbsCs for yuccaols A-E, yuccalide A, and gloriosaols A and C-E extracted from the bark of Y. schidigera by comparing experimental and calculated TDDFT ECD spectra. The absolute configurations of yuccalides B-C and gloriosaol B have not yet been determined.
4.5. Scillascillin-Type Homoisoflavonoids (49-65)
The first homoisoflavonoids with a spiro-structure, scillascillin (49) and 2-hydroxy-7-O-methyl-scillascillin (50), were isolated from the bulbs of Scilla scilloides (Lindl.) Druce by Kouno et al. (1973) (Table 6) [5]. Corsaro et al. (1992) measured Cotton effects for compounds 49 and 51 (2-hydroxy-scillascillin) and assigned their AbsCs as 3R [14]. Adinolfi et al. (1990) determined the AbsC as 3R of the scillascillin-type homoisoflavonoids of Muscari spp., i.e., isomuscomosin (52), muscomosin (53), 3′,4′,5-trihydroxy-7-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (54), 5,5′-dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (55), and 3′,5-dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (56) using several methods [79]. The compounds were first methylated with diazomethane in ether, and 52-54, 56 gave the same dextrorotatory product, namely 5-hydroxy-3′,4′,7-trimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one. This product was then esterified at C-5 with p-bromobenzoyl chloride, followed by reduction with sodium borohydride to give a deoxygenated compound at C-4, which was again p-bromobenzoylated. The RelC and AbsC of the products were determined by NMR and confirmed by measurements of their CD curves and XRD [79]. The AbsC of 55 was assigned based on the measurement of its Cotton effects and X-ray crystallography. Waller et al. (2013) could not directly determine AbsC at C-2 of 2-hydroxy-7-O-methyl-scillascillin (50) due to the instability of the hemiacetal ring [22]. Acetylation of this compound solved this problem by yielding two diastereomers, and their AbsCs were confirmed as 2R,3R and 2S,3R using NMR and ECD experiments by comparing experimental and calculated TDDFT ECD spectra. The AbsC of comp. 57 (5,7,3′-trihydroxy-4′-methoxy-6-methylspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]-trien]-4-one) was determined to be 3R by extensive NMR analysis, based on the similarity of the experimental CD spectra to those of comp. 59 (scillavone A) [27]. In contrast, Nishida et al. (2008) determined the RelC of comp. 59 by XRD and then its AbsC (3R) by comparison of Cotton effects with the literature data [79]. The AbsC of compounds 58 and 60-64 has not yet been determined [15,20,22], although the literature to date indicates that the scillascillin-type homoisoflavonoids described so far have only the 3R configuration.
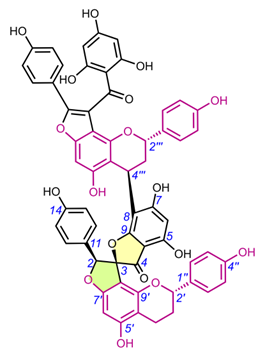
Compound 65 (protosappanin D) belongs to the group of dimeric protosappanin-type homoisoflavonoids, which are characterized by the presence of a biphenyl sappanin moiety forming a methyloxocane ring. It has a very interesting structure with two spiro systems, but its spectral and physicochemical properties have not been reported, not to mention its AbsC. Washiyama et al. (2009) only mentioned that it was extracted from Sappan Lignum (Caesalpinia sappan heartwood) purchased from Uchida Wakanyaku Ltd. [36].
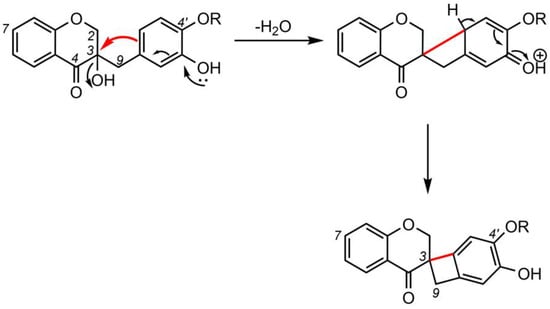
5. Biosynthesis of Spiro-Flavonoids
6. Biological Activities of Spiro-Flavonoids
6.2. Anti-Inflammatory Activity
6.3. Neuroprotective Activity
6.4. Anticancer and Antitumor Activity
6.5. Cytotoxicity/Mutagenicity
6.6. Antiplatelet Activity
6.7. Antidiabetic Activity
6.8. Antibacterial, Antifungal, and Antiviral Activity
6.9. Phytotoxic Activity
6.10. Other Activities
7. Conclusions and Futher Directions
Spiro-flavonoids, due to the presence of an unusual structural element such as spiro-carbon, are attracting increasing interest because of their chemical and biological properties. A total of 65 spiro-flavonoid structures which were monomeric, as well as bi-, tri-, and tetrameric, belonging to several groups differing in the type of polyphenolic units and the way they are combined, were isolated and characterized. Spiro-biflavonoids were the most abundant group, most frequently isolated from the families Pinaceae, Thymelaeaceae, Cupressaceae, and Pentaphylacaceae. In turn, the richest source of spiro-compounds (thirty-four structures) was the Asparagaceae family, from which all known scillascillin-type homoisoflavonoids and all spiro-flavostilbenoids were derived. Most of the oligomeric spiro-flavonoids were isolated from woody plant parts (twigs, bark, and roots), while monomeric scillascillin-type homoisoflavonoids were obtained from bulbs. Methods used to isolate them mainly included classical extraction by maceration at room temperature using pure organic solvents of relatively different polarity and their mixtures, most often with water. The subsequent separation steps also included classical separation techniques based on the difference in solubility in two immiscible liquids (liquid-liquid extraction) as well as the use of column liquid chromatography in normal and reversed-phase systems and gel filtration. However, normal-phase column chromatography was the most widely used technique. As modern chemistry strives to be as "green" as possible, future studies should use extraction methods based on less toxic solvents, such as CO2 supercritical fluid extraction or deep eutectic solvents.
The relative and absolute configurations of the complex structures of spiro-flavonoids, frequently containing multiple chiral carbons (including spiro-carbons), have been determined by a number of spectroscopic techniques, including nuclear magnetic resonance (NMR), electronic circular dichroism (ECD), X-ray diffraction (XRD), and chemical methods using chiral derivatizing agents. NMR and XRD are the methods most commonly used to determine their relative configurations, although an increasing number of cases of the use of quantum mechanical (QM) calculations (e.g., modified DP4+ probability method) are reported in the literature. Empirical methods have been used to assign absolute configuration, including the comparison of Cotton effects between known and newly described compounds. However, this method seems to be far from sufficient for structures with more than one chirality center, so it is necessary to systematically and correctly use QM techniques to predict ECD spectra using time-dependent density-functional theory calculations.
This review also summarizes the topic of possible pathways for spiro-flavonoid biosynthesis. The available knowledge provides some clues pointing towards mechanisms involving radical coupling reactions. Nevertheless, it is noteworthy that there are essentially no works that explore this topic in depth, for example, using isotopic labeling, which we believe should provide a direction for future research.
The potential health benefits of spiro-flavonoids have been summarized and the available results indicate significant anti-inflammatory, neuroprotective, antitumor/anticancer, and antidiabetic properties in vitro and in vivo of some spiro-biflavonoids and spiro-flavostilbenoids. On the other hand, scillascillin-type homoisoflavonoids showed good vasorelaxant activity. Therefore, future research should focus on these aspects of their activity.
Supplementary Materials: The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28145420/s1, Table S1: Biological activity of spiro-flavonoids with information on the models used and results for the substances tested.
12-HETE 12-Hydroxy-5,8,10,14-eicosatetraenoic acid
CDA Chiral derivatizing agent
This entry is adapted from the peer-reviewed paper 10.3390/molecules28145420