The kallikrein–kinin system consists of the two kininogen substrates present in the blood plasma, and two serine proteases: the plasma and tissue kallikreins. The action of the latter on kininogens produces small peptides, the kinins, short-lived, but endowed by powerful pharmacologic actions on blood vessels and other tissues. Several classes of drugs alter kinin formation or action at their receptors for a therapeutic benefit.
- kallikrein–kinin system
- kininogens
- bradykinin
- B1 receptor
- B2 receptor
1. Kallikrein–Kinin Systems: The Formation and Clearance of Kinins
| Abbreviation | Standing for | Corresponding Gene |
|---|---|---|
| ACE | angiotensin-I-converting enzyme | ACE |
| angiopoietin 1 | ANGPT1 | |
| APN | aminopeptidase N | ANPEP |
| Arg-CP | arginine carboxypeptidase | |
| B1R | bradykinin B1 receptor | BDKRB1 |
| B2R | bradykinin B2 receptor | BDKRB2 |
| BK | bradykinin | |
| C1INH | C1-esterase inhibitor | SERPING1 |
| D6 | domain 6 of HK | |
| FXII | coagulation factor XII | F12 |
| FXIIa | activated factor XII | |
| HAE | hereditary angioedema | |
| HAE-C1INH | HAE caused by C1INH haplodeficiency | |
| HK | high-molecular-weight kininogen | KNG1 |
| KKS | Kallikrein–kinin system | |
| KLK-1 | tissue kallikrein | KLK1 |
| LK | low-molecular-weight kininogen | KNG1 |
| Lys-BK | kallidin | |
| mAb | therapeutic monoclonal antibody | |
| NPA | non-peptide antagonist | |
| plasminogen | PLG | |
| tPA | tissue plasminogen activator | PLAT |
| uPA | urokinase-type plasminogen | PLAU |
2. Kinin Receptors
3. Drugs of the KKS in therapeutics
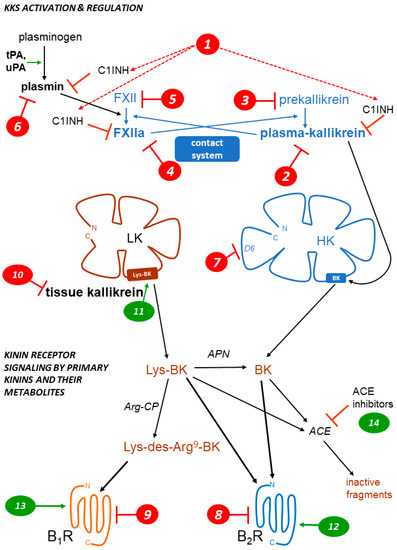
The therapeutic showcase of the KKS is presently hereditary angioedema (HAE), a rare disease most often caused by the haplodeficiency of C1INH: numerous mutations transmitted in an autosomal dominant manner are known in the corresponding gene SERPING1 [32]. HAE is characterized by recurrent episodes (attacks) of swelling due to fluid extravasation; limbs, the orofacial and genital areas, and the intestine can be affected. Attacks may be life-threatening (suffocation), painful and incapacitating. The physio-pathology of HAE and its management have been recently reviewed [32][33][34][35]. While C1INH inhibits several proteases in the contact, fibrinolytic and complement systems, bradykinin is believed to be the ultimate mediator of HAE-C1INH attacks.
Drugs and biotechnological treatments are used or proposed for attack prevention (prophylaxis), to abort attacks (“on demand” treatments), or both. Several HAE therapies that affect the KKS are approved or under development (Table 2). The parenteral administration of C1INH, or gene therapy to increase the hepatic biosynthesis of normal C1INH, is physiologically sound for HAE-C1INH. This approach is supported by multiple clinical trials for C1INH concentrates. The heart of the contact system is also targeted in HAE (Fig. 1, Table 2): plasma kallikrein or its proenzyme prekallikrein, FXIIa or its proenzyme FXII can be suppressed or pharmacologically inhibited by several pharmacological or biotechnological interventions. The proof of concept for a further level of intervention on the contact system has been recently reported in a preclinical study: the mAb 3E8 targets domain 6 (D6) of HK, thus inhibiting the assembly of the trimolecular complex HK-prekallikrein-factor XI (mode of action 7 in Fig. 1). In transgenic mice that express human HK, mAb 3E8 inhibits dextran sulfate-induced BK formation and FXII activation [36].
| Type of Agent Mode of Action Marker in Figure 1 |
Drug or Intervention | Development Status | Ref. |
|---|---|---|---|
| Parenteral replacement of C1INH 1 | various C1INH concentrates, natural or recombinant | approved, widely used | [37] |
| Gene therapy to increase the endogenous synthesis of C1INH 1 | BMN 311 HAE | clinical trials | [38] |
| OTL-105 HAE | preclinical | [39] | |
| Kunitz-domain-based peptide inhibitor of plasma kallikrein 2 | ecallantide | approved | [40] |
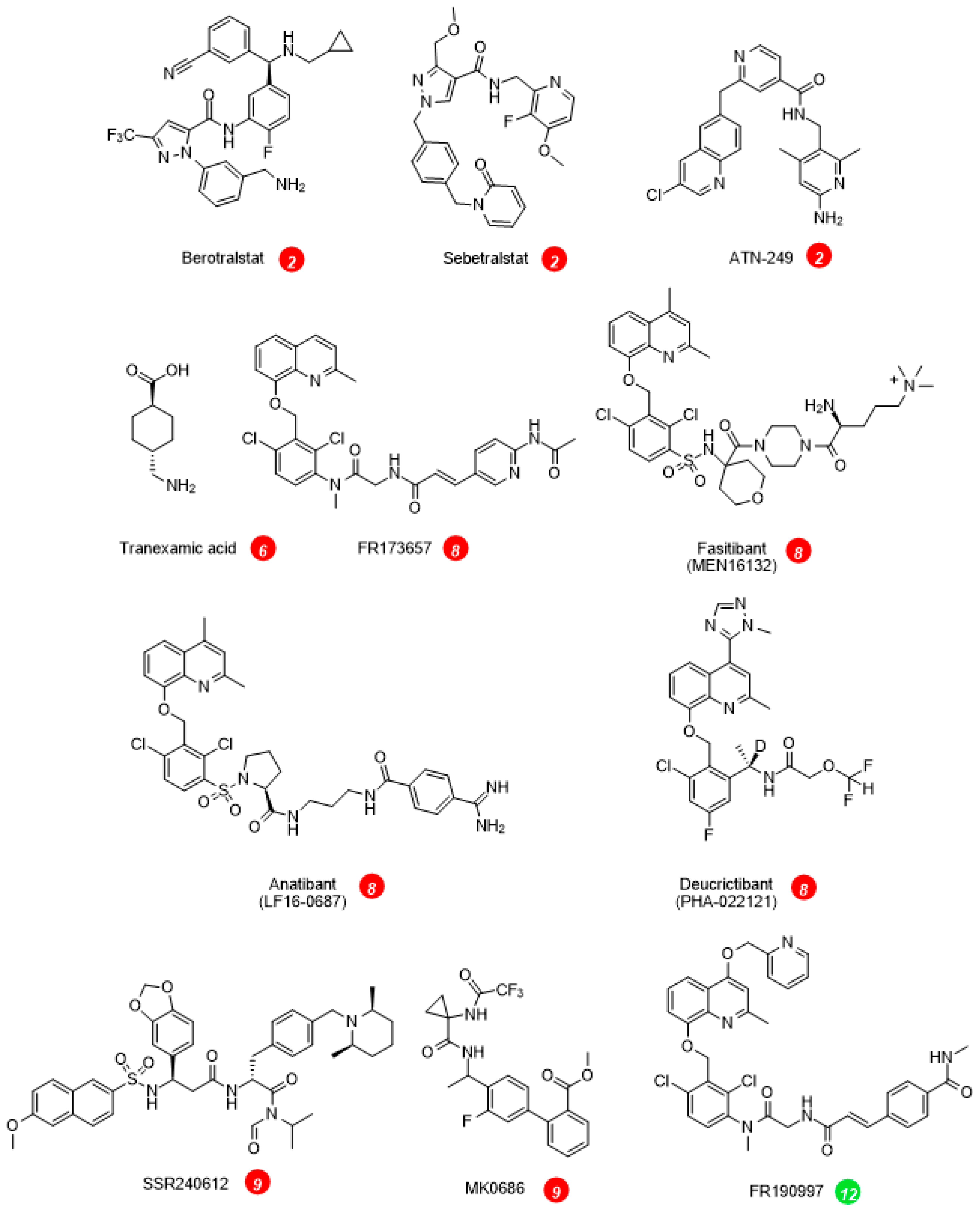
| Small molecule inhibitors of plasma kallikrein 2 | berotralstat (BCX7353) | approved | [41] |
| sebetralstat (KVD-900) | clinical trials | [42] | |
| ATN-249, ATN-111 | clinical trials | [43] | |
| Anti-plasma kallikrein mAb 2 | lanadelumab | approved | [44] |
| STAR-0215 | clinical trials | [45] | |
| Transfer of a gene encoding an anti-plasma kallikrein mAb 2 | RegenxBio undisclosed | preclinical | [46] |
| Antisense suppressor of hepatic plasma prekallikrein production 3 | donidalorsen (PKK-L Rx) | clinical trials | [47] |
| Gene therapy to disrupt hepatic plasma prekallikrein production 3 | NTLA-2002 | clinical trials | [48] |
| Small molecule inhibitor of factor XIIa 4 | KV998086 | preclinical | [49] |
| Anti-factor XII mAb 4 | garadacimab (CSL312) | clinical trials | [50] |
| Small interfering RNA targeting factor XII mRNA 5 | ALN-F12 | preclinical, halted? | [51] |
| ARC-F12 | preclinical, halted? | [52] | |
| Plasmin/tPA inhibitor 6 | tranexamic acid | approved, 2nd line prophylactic agent | [53] |
| Bradykinin B2R antagonists 8 | peptide icatibant | approved | [54] |
| NPA deucrictibant (PHA-022121, PHA-121) | clinical trials | [24][55] |
On the effector side, the BK B2R antagonists inhibit the vascular manifestations of HAE (Table 2, Fig. 1). The injectable and rapidly cleared peptide antagonist icatibant is widely used to abort HAE attacks. The nonpeptide B2R antagonist deucrictibant [24] (Fig. 2) is orally bioavailable, more potent, and longer lived than icatibant in vivo; it is currently developed for on demand treatment of HAE attacks (a potentially convenient substitute to subcutaneous icatibant, Table 2). Chronically administered deucrictibant will also be tested for prophylaxis. Both icatibant and deucrictibant are competitive and reversible an-tagonists at the human B2R [24]. There is clear evidence of fibrinolytic system activation during HAE attacks [56]. Oral tranexamic acid, an inhibitor of plasmin and tissue plasminogen activator, has been approved as a second line prophylactic treatment of HAE.
Other ongoing or terminated therapeutic projects exploited inhibitors of the KKS. Some comments are offered here concerning specific indications. Pain is one of the cardinal signs of inflammation; despite good preclinical evidence, the clinical development of sophisticated and orally bioavailable B1R antagonists, SSR240612 and MK0686 (Fig. 2, mode of action 9 in Fig. 1), has failed due to their lack of efficacy in phase 2 trials (Table 3, Fig. 2) [57]. Fasitibant, a B2R antagonist injected in an intraarticular manner, has also failed to relieve pain associated with knee osteoarthritis (Fig. 2, mode of action 8) [58]. The B1R antagonist BI1026706 (Fig. 2, mode of action 9) failed to prevent diabetic macular edema [59] and the B2R antagonist anatibant (Fig. 2, mode of action 8) was ineffective to prevent post-traumatic cerebral edema [60]. The unsuccessful clinical research concerning the B1R as a druggable target could benefit from the repurposing of potent and specific antagonists that have passed successfully clinical phase 1 development (Fig. 2), for instance for the prevention of COVID-19 complications [61]. An efficient monoclonal antibody that blocks the enzymatic action of tissue kallikrein, DX-2300, has been developed and shown of potential interest in preclinical research [62] (mode of action 10). Other therapeutic investigations of the KKS antagonists are reviewed elsewhere [63].
Whether KKS stimulation can be of therapeutic value is generally a debate at an early stage (modes of action 11 to 14, Fig. 1) [63]. It is already well supported that ACE inhibitors, widely prescribed anti-hypertensive drugs, mediate a part of their beneficial effects via a potentiation of the vasodilator effects of kinins mediated by the B2R [64] (mode of action 14). On the other hand, a nonpeptide and long-acting B2R agonist structurally related to antagonists, FR190997 (Fig. 2, mode of action 12) is clearly pro-inflammatory in animals [65]. Let us mention here the clinical development of tissue kallikrein (KLK-1, mode of action 11). Endogenous tissue kallikrein promotes reparative neovascularization following experimental ischemia and protects the heart in animal models of pathologies [66][67]. This enzyme, produced in a regulated manner in the kidney, is released in urine and protects from sodium overload and salt-sensitive hypertension [68]. Tissue kallikrein also participates to flow-dependent vasodilation, a local circulatory adaptative mechanism [69]. So, why not consider the parenteral administration of tissue kallikrein in therapeutics? In China, active KLK-1 purified from human urine has reached clinical use for acute ischemic stroke. When added to standard thrombolytic therapy, parenteral tissue kallikrein improved the neurological recovery in a significant manner [70]. A pharmaceutically refined recombinant tissue kallikrein, DM199, is being clinically developed for cerebrovascular and renal dysfunctions [71][72].
The medicinal chemistry related to the KKS has reached maturity, with the development of modern drugs, injectable biotechnological proteins, and advanced gene therapy projects. In addition to C1INH replacement therapy, HAE has been the focus of intense drug development efforts based on a limited number of validated targets (plasma kal-likrein, FXIIa and their respective zymogens, the B2R). The recent transition to oral therapies is also noted. Although drug targeting of KKS in animal models provided promising therapeutic leads, disappointing clinical outcomes followed, as in other therapeutic areas. The existence of orally bioavailable drugs that have at least passed clinical phase 1 development (B1R and B2R antagonists, plasma kallikrein inhibitors) could facilitate their repurposing for additional therapeutic indications.
This entry is adapted from the peer-reviewed paper 10.3390/ddc2030028
References
- Leeb-Lundberg, L.M.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77.
- Kaplan, A.P.; Joseph, K.; Ghebrehiwet, B. The complex role of kininogens in hereditary angioedema. Front. Allergy 2022, 3, 952753.
- Kaplan, A.P. Enzymatic pathways in the pathogenesis of hereditary angioedema: The role of C1 inhibitor therapy. J. Allergy Clin. Immunol. 2010, 126, 918–925.
- Gauberti, M.; Potzeha, F.; Vivien, D.; Martinez de Lizarrondo, S. Impact of Bradykinin Generation During Thrombolysis in Ischemic Stroke. Front. Med. 2018, 5, 195.
- Dobó, J.; Major, B.; Kékesi, K.A.; Szabó, I.; Megyeri, M.; Hajela, K.; Juhász, G.; Závodszky, P.; Gál, P. Cleavage of kininogen and subsequent bradykinin release by the complement component: Mannose-binding lectin-associated serine protease (MASP)-1. PLoS ONE 2011, 6, e20036.
- Charest-Morin, X.; Hébert, J.; Rivard, G.É.; Bonnefoy, A.; Wagner, E.; Marceau, F. Comparing Pathways of Bradykinin Formation in Whole Blood From Healthy Volunteers and Patients With Hereditary Angioedema Due to C1 Inhibitor Deficiency. Front. Immunol. 2018, 9, 2183.
- Verhoef, J.J.F.; Barendrecht, A.D.; Nickel, K.F.; Dijkxhoorn, K.; Kenne, E.; Labberton, L.; McCarty, O.W.T.; Schiffelers, R.; Heijnen, H.; Hendricks, A.P.; et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood 2017, 129, 1707–1717.
- Yousef, G.M.; Chang, A.; Scorilas, A.; Diamandis, E.P. Genomic organization of the human kallikrein gene family on chromosome 19q13.3-q13.4. Biochem. Biophys. Res. Commun. 2000, 276, 125–133.
- David Deperthes; François Marceau; Gilles Frenette; Claude Lazure; Roland R Tremblay; Jean Y Dubé; Human kallikrein hK2 has low kininogenase activity while prostate-specific antigen (hK3) has none. Biochimica Et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1997, 1343, 102-106, .
- Charest-Morin, X.; Raghavan, A.; Charles, M.L.; Kolodka, T.; Bouthillier, J.; Jean, M.; Robbins, M.S.; Marceau, F. Pharmacological effects of recombinant human tissue kallikrein on bradykinin B2 receptors. Pharmacol. Res. Perspect. 2015, 3, e00119.
- Marceau, F.; Rivard, G.E.; Gauthier, J.M.; Binkley, K.E.; Bonnefoy, A.; Boccon-Gibod, I.; Bouillet, L.; Picard, M.; Levesque, G.; Elfassy, H.L.; et al. Measurement of Bradykinin Formation and Degradation in Blood Plasma: Relevance for Acquired Angioedema Associated With Angiotensin Converting Enzyme Inhibition and for Hereditary Angioedema Due to Factor XII or Plasminogen Gene Variants. Front. Med. 2020, 7, 358.
- Marceau, F.; Bachelard, H.; Charest-Morin, X.; Hébert, J.; Rivard, G.E. In vitro modeling of bradykinin-mediated angioedema states. Pharmaceuticals 2020, 13, 201.
- Fryer, R.M.; Segreti, J.; Banfor, P.N.; Widomski, D.L.; Backes, B.J.; Lin, C.W.; Ballaron, S.J.; Cox, B.F.; Trevillyan, J.M.; Reinhart, G.A.; et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: Rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–955.
- Ishida, H.; Scicli, A.G.; Carretero, O.A. Contributions of various rat plasma peptidases to kinin hydrolysis. J. Pharmacol. Exp. Ther. 1989, 251, 817–820.
- Defendi, F.; Charignon, D.; Ghannam, A.; Baroso, R.; Csopaki, F.; Allegret-Cadet, M.; Ponard, D.; Favier, B.; Cichon, S.; Nicolie, B.; et al. National Reference Centre for Angioedema CREAK. Enzymatic assays for the diagnosis of bradykinin-dependent angioedema. PLoS ONE 2013, 8, e70140.
- Marceau, F.; Rivard, G.E.; Hébert, J.; Gauthier, J.; Bachelard, H.; Gangnus, T.; Burckhardt, B.B. Picomolar Sensitivity Analysis of Multiple Bradykinin-Related Peptides in the Blood Plasma of Patients With Hereditary Angioedema in Remission: A Pilot Study. Front. Allergy 2022, 3, 837463.
- Cugno, M.; Salerno, F.; Nussberger, J.; Bottasso, B.; Lorenzano, E.; Agostoni, A. Bradykinin in the ascitic fluid of patients with liver cirrhosis. Clin. Sci. 2001, 101, 651–657.
- Hofman, Z.L.M.; van den Elzen, M.T.; Kuijpers, J.; de Maat, S.; Hack, C.E.; Knulst, A.C.; Röckmann, H.; Maas, C. Evidence for bradykinin release in chronic spontaneous urticaria. Clin. Exp. Allergy 2020, 50, 343–351.
- Mostmans, Y.; De Smedt, K.; Richert, B.; Elieh Ali Komi, D.; Maurer, M.; Michel, O. Markers for the involvement of endothelial cells and the coagulation system in chronic urticaria: A systematic review. Allergy 2021, 76, 2998–3016.
- Barratt-Due, A.; Johansen, H.T.; Sokolov, A.; Thorgersen, E.B.; Hellerud, B.C.; Reubsaet, J.L.; Seip, K.F.; Tønnessen, T.I.; Lindstad, J.K.; Pharo, A.; et al. The role of bradykinin and the effect of the bradykinin receptor antagonist icatibant in porcine sepsis. Shock 2011, 36, 517–523.
- Sparkenbaugh, E.M.; Kasztan, M.; Henderson, M.W.; Ellsworth, P.; Davis, P.R.; Wilson, K.J.; Reeves, B.; Key, N.S.; Strickland, S.; McCrae, K.; et al. High molecular weight kininogen contributes to early mortality and kidney dysfunction in a mouse model of sickle cell disease. J. Thromb. Haemost. 2020, 18, 2329–2340.
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46.
- Hock, F.J.; Wirth, K.; Albus, U.; Linz, W.; Gerhards, H.J.; Wiemer, G.; Henke, S.; Breipohl, G.; König, W.; Knolle, J.; et al. Hoe 140 a new potent and long acting bradykinin-antagonist: In vitro studies. Br. J. Pharmacol. 1991, 102, 769–773.
- Lesage, A.; Marceau, F.; Gibson, C.; Loenders, B.; Katzer, W.; Ambrosi, H.D.; Saupe, J.; Faussner, A.; Pardali, E.; Knolle, J. In vitro pharmacological profile of PHA-022121, a small molecule bradykinin B2 receptor antagonist in clinical development. Int. Immunopharmacol. 2022, 105, 108523.
- Marceau, F.; Bachelard., H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305.
- Larrivée, J.-F.; Bachvarov, D.R.; Houle, F.; Landry, J.; Huot, J.; Marceau, F. Role of the mitogen-activated protein kinases in the expression of the kinin B1 receptors induced by tissue injury. J. Immunol. 1998, 160, 1419–1426.
- Moreau, M.E.; Bawolak, M.T.; Morissette, G.; Adam, A.; Marceau, F. Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells. Mol. Pharmacol. 2007, 71, 949–956.
- Koumbadinga, G.A.; Désormeaux, A.; Adam, A.; Marceau, F. Effect of interferon-γ on inflammatory cytokine-induced bradykinin B1 receptor expression in human vascular cells. Eur. J. Pharmacol. 2010, 647, 117–125.
- Oliveira, A.C.; Vicentino, A.R.R.; Andrade, D.; Pereira, I.R.; Saboia-Vahia, L.; Moreira, O.D.C.; Carvalho-Pinto, C.E.; Mota, J.B.D.; Maciel, L.; Vilar-Pereira, G.; et al. Genetic Ablation and Pharmacological Blockade of Bradykinin B1 Receptor Unveiled a Detrimental Role for the Kinin System in Chagas Disease Cardiomyopathy. J. Clin. Med. 2023, 12, 2888.
- Emanueli, C.; Bonaria Salis, M.; Stacca, T.; Pintus, G.; Kirchmair, R.; Isner, J.M.; Pinna, A.; Gaspa, L.; Regoli, D.; Cayla, C.; et al. Targeting kinin B1 receptor for therapeutic neovascularization. Circulation 2002, 105, 360–366.
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852.
- Remy S. Petersen; Lauré M. Fijen; Marcel Levi; Danny M. Cohn; Hereditary Angioedema: The Clinical Picture of Excessive Contact Activation. Seminars in Thrombosis and Hemostasis 2022, in press, 1-11, .
- Anna Valerieva; Hilary J. Longhurst; Treatment of hereditary angioedema—single or multiple pathways to the rescue. Frontiers in Allergy 2022, 3, 952233, .
- Marcus Maurer; Markus Magerl; Stephen Betschel; Werner Aberer; Ignacio J. Ansotegui; Emel Aygören-Pürsün; Aleena Banerji; Noémi-Anna Bara; Isabelle Boccon-Gibod; Konrad Bork; et al. The international WAO/EAACI guideline for the management of hereditary angioedema – The 2021 revision and update. World Allergy Organization Journal 2022, 15, 100627, .
- Konrad Bork; Karin Wulff; Günther Witzke; Petra Staubach; Jochen Hardt; Peter Meinke; Gene Mutations Linked to Hereditary Angioedema in Solitary Angioedema Patients With Normal C1 Inhibitor. The Journal of Allergy and Clinical Immunology. In Practice 2023, in press, 1-9, .
- Zu-Lin Chen; Pradeep K. Singh; Katharina Horn; Marissa R. Calvano; Shigeru Kaneki; Keith R. McCrae; Sidney Strickland; Erin H. Norris; Anti-HK antibody inhibits the plasma contact system by blocking prekallikrein and factor XI activation in vivo. Blood Advances 2023, 7, 1156-1167, .
- Longhurst, H.; Farkas, H. Biological therapy in hereditary angioedema: Transformation of a rare disease. Expert Opin. Biol. Ther. 2020, 20, 493–501.
- Biomarin. Available online: https://www.biomarin.com/our-treatments/pipeline/bmn-331-for-hae/ (accessed on 1 May 2023).
- Pharming Group N.V. Available online: https://www.pharming.com/pipeline (accessed on 1 May 2023).
- Duffey, H.; Firszt, R. Management of acute attacks of hereditary angioedema: Role of ecallantide. J. Blood Med. 2015, 6, 115–123.
- Ahuja, M.; Dorr, A.; Bode, E.; Boulton, A.P.R.; Buckland, M.; Chee, S.; Dalley, C.; Denman, S.; Ekbote, A.; Elkhalifa, S.; et al. Berotralstat for the prophylaxis of hereditary angioedema-Real-world evidence data from the United Kingdom. Allergy 2023, 78, 1380–1383.
- Aygören-Pürsün, E.; Zanichelli, A.; Cohn, D.M.; Cancian, M.; Hakl, R.; Kinaciyan, T.; Magerl, M.; Martinez-Saguer, I.; Stobiecki, M.; Farkas, H.; et al. An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: A two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial. Lancet 2023, 401, 458–469.
- Kalfus, I.; Offman, E.; McDonald, A. Pharmacokinetics, safety, and potency of ATN-249, a novel oral plasma kallikrein inhibitor for hereditary angioedema. Allergy Asthma Clin. Immunol. 2019, 15 (Suppl. S4), 45.
- Riedl, M.A.; Maurer, M.; Bernstein, J.A.; Banerji, A.; Longhurst, H.J.; Li, H.H.; Lu, P.; Hao, J.; Juethner, S.; Lumry, W.R.; et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy 2020, 75, 2879–2887.
- Astria Therapeutics. Available online: https://astriatx.com/our-science/scientific-presentations-and-publications/ (accessed on 26 April 2023).
- REGENXBIO Inc. Available online: https://ir.regenxbio.com/news-releases/news-release-details/regenxbio-reports-continued-progress-across-programs-year-end-0/ (accessed on 1 May 2023).
- Ferrone, J.D.; Bhattacharjee, G.; Revenko, A.S.; Zanardi, T.A.; Warren, M.S.; Derosier, F.J.; Viney, N.J.; Pham, N.C.; Kaeser, G.E.; Baker, B.F.; et al. IONIS-PKKRx a Novel Antisense Inhibitor of Prekallikrein and Bradykinin Production. Nucleic Acid. Ther. 2019, 29, 82–91.
- Intellia Therapeutics, Inc. Available online: https://www.intelliatx.com/our-science/publications-and-presentations/ (accessed on 26 April 2023).
- KalVista Pharmaceuticals. Available online: https://www.kalvista.com/products-pipeline/factor-xiia (accessed on 26 April 2023).
- Craig, T.J.; Reshef, A.; Li, H.H.; Jacobs, J.S.; Bernstein, J.A.; Farkas, H.; Yang, W.H.; Stroes, E.S.G.; Ohsawa, I.; Tachdjian, R.; et al. Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): A global, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1079–1090.
- Liu, J.; Qin, J.; Borodovsky, A.; Racie, T.; Castoreno, A.; Schlegel, M.; Maier, M.A.; Zimmerman, T.; Fitzgerald, K.; Butler, J.; et al. An investigational RNAi therapeutic targeting Factor XII (ALN-F12) for the treatment of hereditary angioedema. RNA 2019, 25, 255–263.
- Arrowhead Pharmaceuticals, Inc. Available online: http://ir.arrowheadpharma.com/news-releases/news-release-details/arrowhead-pharmaceuticals-presents-new-data-arc-f12-and-arc-lpa (accessed on 1 May 2023).
- Wintenberger, C.; Boccon-Gibod, I.; Launay, D.; Fain, O.; Kanny, G.; Jeandel, P.Y.; Martin, L.; Gompel, A.; Bouillet, L. Tranexamic acid as maintenance treatment for non-histaminergic angioedema: Analysis of efficacy and safety in 37 patients. Clin. Exp. Immunol. 2014, 178, 112–117.
- Maurer, M.; Aberer, W.; Caballero, T.; Bouillet, L.; Grumach, A.S.; Botha, J.; Andresen, I.; Longhurst, H.J.; IOS Study Group. The Icatibant Outcome Survey: 10 years of experience with icatibant for patients with hereditary angioedema. Clin. Exp. Allergy 2022, 52, 1048–1058.
- Maurer, M.; Anderson, J.; Aygören-Pürsün, E.; Bouillet, L.; Baeza, M.L.; Chapdelaine, H.; Cohn, D.; Du-Thanh, A.; Fain, O.; Farkas, H.; et al. Efficacy And Safety of Bradykinin B2 Receptor Inhibition With Oral PHVS416 In Treating Hereditary Angioedema Attacks: Results Of RAPIDe-1 Phase 2 Trial. J. Allergy Clin. Immunol. 2023, 151, AB134.
- A. Reshef; A. Zanichelli; H. Longhurst; A. Relan; C. E. Hack; Elevated D ‐dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy 2015, 70, 506-513, .
- Christopher I Fincham; Alessandro Bressan; Marielle Paris; Cristina Rossi; Daniela Fattori; Bradykinin receptor antagonists – a review of the patent literature 2005 – 2008. Expert Opinion on Therapeutic Patents 2009, 19, 919-941, .
- Werner CG, Pavelka K, Nizzardo A, Rossi C, Scartoni S, Contini MP, di Molfetta S, Bertolotti M, Capriati A, Maggi CA. A Double-Blind, Randomized, Controlled, Four parallel Arm, Dose-Finding Study to Evaluate the Efficacy, Safety, Tolerability, and Pharmacokinetics of Single Intra-Articular (IA) Injections of Fasitibant in Patients with Symptomatic OA of the Knee [abstract]. Arthritis Rheumatol. 2015, 67 (suppl 10). https://acrabstracts.org/abstract/a-double-blind-randomized-controlled-four-parallel-arm-dose-finding-study-to-evaluate-the-efficacy-safety-tolerability-and-pharmacokinetics-of-single-intra-articular-ia-injections-of-fas/. [Last Accessed 1 May 2023].
- Gabriele E. Lang; Ramin Tadayoni; Wenbo Tang; Claudia Barth; Cornelia Weiss-Haljiti; Victor Chong; on behalf of the BI 1026706 Study Group; Bradykinin 1 Receptor Antagonist BI1026706 Does Not Reduce Central Retinal Thickness in Center-Involved Diabetic Macular Edema. Translational Vision Science & Technology 2020, 9, 25-25, .
- Haleema Shakur; Peter Andrews; Toomas Asser; Laura Balica; Cristian Boeriu; Juan Diego Ciro Quintero; Yashbir Dewan; Patrick Druwé; Olivia Fletcher; Chris Frost; et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 2009, 10, 109, .
- Gabriel Moreira de M Mendes; Israel Júnior Borges Do Nascimento; Paulo Hs. Marazzi-Diniz; Izabela B. Da Silveira; Matheus F. Itaborahy; Luiz E. Viana; Filipe A. Silva; Monique F Santana; Rebecca Aa. Pinto; Bruna G. Dutra; et al. The des-Arg9-bradykinin/B1R axis: Hepatic damage in COVID-19. Frontiers in Physiology 2022, 13, 1080837, .
- Daniel J. Sexton; Ting Chen; Diana Martik; Petr Kuzmic; Guannan Kuang; Jie Chen; Andrew E. Nixon; Bruce L. Zuraw; Rosanna M. Forteza; William M. Abraham; et al. Specific inhibition of tissue kallikrein 1 with a human monoclonal antibody reveals a potential role in airway diseases. null 2009, 422, 383-392, .
- François Marceau; Drugs of the Kallikrein–Kinin System: An Overview. Drugs and Drug Candidates 2023, 2, 538-553, .
- James V. Gainer; Jason D. Morrow; Angela Loveland; Debbie J. King; Nancy J. Brown; Effect of Bradykinin-Receptor Blockade on the Response to Angiotensin-Converting–Enzyme Inhibitor in Normotensive and Hypertensive Subjects. The New England Journal of Medicine 1998, 339, 1285-1292, .
- Izumi Hayashi; Keiko Ishihara; Yuji Kumagai; Masataka Majima; Proinflammatory characteristics of a nonpeptide bradykinin mimic, FR190997, in vivo. British Journal of Pharmacology 2001, 133, 1296-1306, .
- Oliver A. Stone; Christine Richer; Costanza Emanueli; Vincent van Weel; Paul H.A. Quax; Rajesh Katare; Nicolle Kraenkel; Paola Campagnolo; Luciola S. Barcelos; Mauro Siragusa; et al. Critical Role of Tissue Kallikrein in Vessel Formation and Maturation. Arteriosclerosis, Thrombosis, and Vascular Biology 2009, 29, 657-664, .
- Matthias Koch; Frank Spillmann; Andreas Dendorfer; Dirk Westermann; Christine Altmann; Merdad Sahabi; Sophie Van Linthout; Michael Bader; Thomas Walther; Heinz-Peter Schultheiss; et al. Cardiac function and remodeling is attenuated in transgenic rats expressing the human kallikrein-1 gene after myocardial infarction. European Journal of Pharmacology 2006, 550, 143-148, .
- Katori, M.; Majima, M.; Renal (tissue) kallikrein-kinin system in the kidney and novel potential drugs for salt-sensitive hypertension.. Prog. Drug Res. 2014, 69, 59-109, .
- Sonia Bergaya; Pierre Meneton; May Bloch-Faure; Eric Mathieu; François Alhenc-Gelas; Bernard I. Lévy; Chantal M. Boulanger; Decreased Flow-Dependent Dilation in Carotid Arteries of Tissue Kallikrein–Knockout Mice. Circulation Research 2001, 88, 593-599, .
- Jing Wu; Le Wang; Jinmin Liu; Urinary Kallidinogenase plus rt-PA Intravenous Thrombolysis for Acute Ischemic Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Computational and Mathematical Methods in Medicine 2022, 2022, 1500669, .
- Michelle Alexander-Curtis; Rick Pauls; Julie Chao; John J Volpi; Philip M Bath; Todd A. Verdoorn; Human tissue kallikrein in the treatment of acute ischemic stroke. Therapeutic Advances in Neurological Disorders 2019, 12, 1756286418821918, .
- Faheem Shehjar; Briana Maktabi; Zainab A. Rahman; Ghaith A. Bahader; Antonisamy William James; Ahmed Naqvi; Reetika Mahajan; Zahoor A. Shah; Stroke: Molecular mechanisms and therapies: Update on recent developments. Neurochemistry International 2023, 162, 105458, .