Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
One of the most remarkable advancements in medical treatments of corneal diseases in recent decades has been corneal transplantation. Corneal defects and diseases are one of the leading causes of blindness globally; therefore, there is a need for gene-based interventions that may mitigate some of these challenges and help reduce the burden of blindness. Corneas being immune-advantaged, uniquely avascular, and transparent is ideal for gene therapy approaches.
- corneal dystrophies
- corneal neovascularization
- gene therapy
1. Introduction
Corneal diseases are one of the leading causes of visual impairment globally [1]. A delicate physiological and functional balance is responsible for the transparency and clarity of the cornea. Therefore, any disease, acquired or genetic, that compromises this state of homeostasis can eventually lead to vision loss. Inherited, acquired, and iatrogenic corneal diseases, including corneal dystrophies, neovascularization, corneal scarring, haze, dry eyes, keratoconus, and corneal injuries, affect normal vision.
Approval of a gene therapy for retinal disease [2] has paved the way for corneal gene therapy; this method continues to develop and rapidly advance, and has a high potential for human application. Corneal gene therapy initially emerged in 1994, when corneal tissues were transduced successfully using replication-deficient adenovirus to treat acquired inflammatory disease [3]. Gene therapy for retinal diseases has made much greater progress when compared to corneal diseases. However, advancements made in the last two decades in the field of corneal gene therapy have brought this form of therapy much closer to clinical trials and application in human patients. Inherited corneal diseases, such as epithelial, stromal, and endothelial dystrophies [4][5], are natural candidates for corneal gene therapy. Complex conditions of multifactorial etiology, such as keratoconus, also have a genetic component associated to it, making it suitable for gene therapy [6].
The anatomic location of corneal epithelium makes it particularly attractive for non-invasive treatment by direct topical instillation of the gene delivery system [7]. Surgical, mechanical, chemical, and electric methods can also be used to administer gene therapy in the cornea. Furthermore, estimating the effectiveness and safety of the corneal treatment is easier due to the rapid and non-invasive visual observation using standard ophthalmologic methods [8]. Since the cornea is avascular and the eye has an altered immunologic profile in the intraocular compartments that usually dampens immune responses, the chances of systemic or even local immune reactions is lower.
The most striking advancement in the treatment of corneal diseases has been corneal transplantation and tissue engineering [9][10]. One of the most transplanted tissues worldwide is the cornea. In penetrating keratoplasty, the entire cornea is replaced, whereas in lamellar keratoplasty, only the damaged layers are replaced with requisite layers of donor tissue [10][11][12]. Unfortunately, in a subset of subjects, corneal transplantation has been associated with poor outcomes due to graft rejection and graft failure. Furthermore, the WHO has reported a shortage of corneal donors, due to which 10–15% of patients are reported to remain untreated [1][13]. Additionally, it has been reported that almost 53% of the world’s population lacks access to corneal transplantation, due to which there is only one cornea available for every seventy needed [14]. Due to the shortage of donor tissue and the fear of transmissible disease [14][15][16], there has been a continuous evolution of approaches in reviving corneal function and vision. An advantage of the cornea is its stability ex-vivo; it can be maintained in artificial physiological environment for weeks. This is especially advantageous for experimental purposes as well as for the administration of treatments prior to corneal transplantation, aiding in reduced chance of graft rejection [17][18].
To reduce global blindness, the development of a tissue-targeted gene-based intervention based on our understanding of molecular mechanisms that can be targeted by gene therapy is of paramount importance. Many of the diseases could be potentially treated using gene therapy by supplying a functional gene or changing the expression levels of specific genes in affected cellular layers. However, the majority of the studies in the field have been focused on the modulation of acquired corneal medical conditions. Regulation of the corneal microenvironment could be achieved using corneal gene therapy in different disorders by using induction or knock down of respective proteins. To improve the efficacy and safety of the treatment, local corneal gene delivery using a variety of vector and delivery modalities can be used to achieve low and continuous concentrations of the proteins [19]. Therefore, delivery of various immune modulators and growth factors to the cornea, in turn affecting local immune responses, inflammation, and proliferation, can be achieved without any systemic side effects. Corneal gene therapy has therefore been applied to the treatment of fibrotic disorders and corneal haze post-refractive procedures. Topical gene delivery to the cornea has the potential to effectuate local protein expression at concentrations unachievable by systemic administration of the transgenes or recombinant proteins [20]. Corneal gene therapy approaches can therefore enhance survival of corneal grafts and improve corneal diseases that currently require a corneal transplantation, thus preventing the need for an allograft [21].
2. Genes of Corneal Diseases
One of the first requirements for a successful gene-based treatment approach for the cornea is to identify the genes that need to be augmented, replaced, or edited, or identification of the pathways to be targeted by the protein product of the selected transgene.
2.1. Genes and Genetics of Inherited Diseases
Corneal dystrophies: Corneal dystrophies represent a heterogenous group of inherited corneal diseases that affect corneal development and cellular function. Corneal dystrophies are classified based on the layer of cornea that they affect. The human cornea essentially consists of the epithelium, the stroma, and the endothelium layer. Bowman’s layer, an acellular structure with densely compacted collagen fibrils, separates the epithelium and the anterior stroma. The Descemet’s membrane is the basement membrane of the endothelial layer that separates the posterior part of the stroma from the endothelium. Corneal dystrophies are classified into four main categories: epithelial and subepithelial dystrophies; Bowman’s layer dystrophies; stromal dystrophies; and Descemet’s membrane and endothelial dystrophies (Figure 1). Each of these dystrophies exhibit distinct clinical features, variable inheritance, age of onset, and course of progression [4][5]. Corneal dystrophies are inherited as autosomal dominant, autosomal recessive, or X-linked.
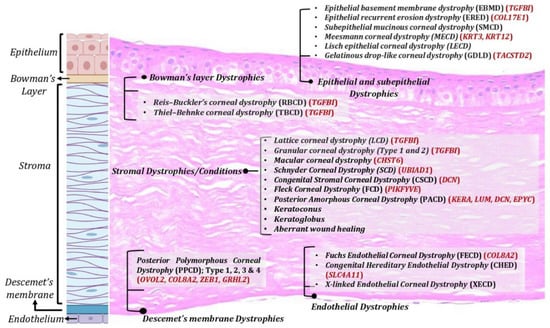
Figure 1. Corneal dystrophies across various corneal layers. Corneal dystrophies affect different layers of the cornea including epithelium, Bowman’s layer, stroma, and Descemet’s membrane and endothelium.
TGFβI-associated dystrophies have been attributed to various mutants in TGFβI gene, with positions 124 and 555 being the most common. Deposition of TGFβ-induced (TGFβ) protein aggregates in the stroma or Bowman layer have been associated with these dystrophies [22]. Several uncommon mutations are scattered across five of the gene’s 17 exons and are associated with divergent clinical presentations of the dystrophy, including Reis–Bucklers corneal dystrophy, Thiel–Behnke corneal dystrophy, lattice corneal dystrophy type 1, granular corneal dystrophy type 1, and granular corneal dystrophy type 2. Epithelial recurrent erosion dystrophy (ERED) is caused by mutation in the COL17A1 gene [23]. Until the discovery of this mutation, Thiel–Behnke corneal dystrophy was thought to result only from mutations in the TGFBI gene. Heterozygous mutations in the KRT3 or KRT12 genes, encoding corneal specific Keratins 3 and 12, respectively [24], have been associated with Meesmann epithelial corneal dystrophy (MECD), a rare autosomal dominant inherited disease. Malfunction of K3 and K12 has been shown to cause mechanical fragility of the anterior corneal epithelium [25]. Various studies have identified at least 24 mutations for MECD, most of which are missense point mutation [24][26][27][28]. Early onset Fuchs endothelial corneal dystrophy (FECD) has been associated with mutations in the COL8A2, SLC4A11, ZEB1, and LOXHD1 genes. The majority of FECD cases are caused by a trinucleotide repeat expansion in the TCF4 gene [29], leading to altered mRNA processing due to sequestration of splicing factor proteins (MBNL1 and MBNL2) to the nuclear RNA foci.
Other inherited diseases: Other inherited disorders affecting the cornea include Aniridia and Mucopolysaccharidosis (MPS). Aniridia exhibits a dominant autosomal inheritance pattern with variable expression in several members of a family. A diverse set of mutations leading to haploinsufficiency of the PAX6 gene, which is expressed in various regions of the eye including the cornea [30], have been demonstrated to be associated this disorder. MPS is a group of inherited metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes that are responsible for the degradation of glycosaminoglycans. Lysosomal accumulation of undegraded glycosaminoglycans in keratocytes causes corneal clouding. MPS demonstrate a high range of clinical manifestations and are classified based on the enzyme that is dysregulated. Although common to most types of MPS (I-IX), corneal manifestations are most common in MPS I, VI, and VII. MPS I is a monogenetic disease caused by loss of function mutations in both copies of the IDUA gene. MPS VII, or Sly syndrome, is caused by mutation in the enzyme β-glucuronidase [31]; MPS VI, or Maroteaux–Lamy syndrome, is caused by mutations leading to dysfunction of the enzyme aryl sulfatase B, causing corneal clouding [32].
2.2. Genes Associated with Acquired Corneal Conditions
2.2.1. Corneal Wound Healing
Disparate injuries can cause fibrotic scarring. Irrespective of the cause of the injury, transforming growth factor β (TGFβ) has been associated with aberrant corneal healing responses. Previous studies have demonstrated the interaction of the TGF-β superfamily proteins with the Smad family to activate downstream signaling [33]. Bone morphogenic protein is a part of TGF-β superfamily that appears to modulate keratocyte proliferation, differentiation, and apoptosis, thereby playing an essential role during corneal wound healing [34]. Studies have shown the important role of BMP7 in the development of the mammalian kidney and eye [35]. The complex wound healing signaling network involves the binding of BMP7 to type I and II receptors, allowing regulation of receptor-regulated Smads (Smad 1, 5, and 8) and inhibitory Smads (Smad 6 and 7) [36]. Improved healing and lessened scarring by overexpression of the inhibitory protein Smad7, thereby causing inhibition of TGFβ signaling pathway, have been observed in animal models [37]. Additionally, hepatocyte growth factor (HGF) and its receptor (c-Met receptor tyrosine kinase) have also been shown to play important roles in normal and healing corneal epithelium and keratocytes in the anterior stroma in vivo [38]. Upregulation of HGF in keratocytes has been observed upon injury to the corneal epithelium [39].
2.2.2. Corneal Neovascularization
Corneal neovascularization occurs in various corneal pathologies, including inflammatory diseases, congenital diseases, contact lens-related hypoxia, inflammatory diseases, autoimmune diseases, chemical burns, trauma, corneal graft rejection, and infectious keratitis [40], which may lead to significant visual impairment or blindness [41]. Various angiogenic factors, such as VEGF, basic fibroblast growth factor (bFGF), matrix metalloproteinase (MMP), platelet-derived growth factors (PDGFs), and interleukin-1 (IL-1), mediate corneal neovascularization. In the retina and the cornea, VEGF-A is the predominant VEGF member driving pathologic neovascularization and is therefore a potential target of several drugs. The VEGF-A binds to two members of receptor tyrosine kinase family, VEGF receptors VEGFR1 and VEGFR2, also known as Flt-1 and KDR, respectively. Vascular endothelial cells (VECs) significantly express both of these receptors, and their expression increases in the presence of inflammation. It has been found that their activation promotes vascular leakage, VEC liberation, and VEC proteolysis [42][43]. Targeting these factors and inhibiting their expression or increasing expression of anti-angiogenic factors may help inhibit angiogenesis. Bevacizumab and ranibizumab are currently used in clinical settings to inhibit VEGF-A signaling pathways [44]. When mixed with non-liposomal lipid and delivered by means of subconjunctival injection, the brain-specific angiogenesis inhibitor 1 (BAI1-ECR) gene showed an effective reduction of corneal neovascularization [45]. Several of these targets that mediate neovascularization, including Flt23k, Flt-1, PEDF, VEGFR Flt-1, MMP-9, and vasohibin-1 [46][47][48][49][50][51], could serve as a potential target candidates for corneal gene therapy.
2.2.3. Corneal Graft Survival
Immunological rejection has always been one of the primary causes for corneal graft rejection [52]. Several different groups have shown significant prolongation of corneal allograft survival using various different transgenes and vectors in a variety of animal models [53]. Several of these transgenes lower immune response [54][55][56][57][58] and reduce angiogenesis, thereby preventing likelihood of donor graft tissue rejection due to inflammation, corneal scarring, and edema [59]. The number of transgenes that have shown success in modulating corneal graft rejection indicates the complex nature of the process and the multiple pathways involved. Prevention of activation of T cells by gene transfer of Cytotoxic T-Lymphocyte Antigen 4 protein (CTLA4-Ig) has shown to effectively prolong graft survival [56][60]. Furthermore, knocking down neuropilin-2 through RNA interference (RNAi) [61] has been shown to reduce the amount of free vascular endothelial growth factor-A (VEGF-A) [62], leading to a decrease in activated T-cell influx and improvement in graft survival.
2.2.4. Multifactorial and Polygenic Diseases
A variety of conditions are associated with reduced central corneal thickness (CCT) which include keratoconus, keratoglobus, brittle cornea syndrome, Ehlers–Danlos syndrome, osteogenesis imperfecta, and myopia [63]. CCT reduction is also associated with various genetic determinants in the context of these diseases, although various systemic and environmental factors are also involved in disease. The most common among these conditions is keratoconus (KC), a complex multifactorial disease associated with a wide variety of etiological factors such as genetic predisposition, environmental insults, oxidative stress, and mechanical injuries or eye rubbing. Alterations in several biochemical mediators have been associated to KC pathogenesis, including lysyl oxidase (LOX) [64][65], MMP2 [66], MMP9 [65][67], and various collagens, including I, III, IV, V, VI, and VII [68]. Familial genetic studies and genome-wide studies of the KC families have identified genomic heterogeneity in the genomic loci among these families [69][70][71][72][73]. Mutations in DOCK9, FLG, TGFβI, SOD1, ZEB1, and VSX1 genes were also found to be responsible for KC prognosis in some selected populations around the world [74][75][76][77][78][79].
3. Gene Therapy for Corneal Diseases
Gene therapy is a technique in which replacing or inactivating (knocking out) the mutated gene occurs as a result of targeted therapeutic delivery of correct nucleic acid directly into the patient’s cells. Gene therapy to treat any inherited corneal dystrophies works via three approaches: (a) Gene inactivation or silencing of the mutated gene that manifests a toxic effect on the cells; (b) Gene correction or replacement of the mutated gene with a normal copy of a healthy gene; and (c) Addition of a healthy copy of the gene that will ectopically express the therapeutic protein to rescue the disease phenotype. Gene therapy has a growing potential in the field of corneal dystrophies due to three major characteristics: accessibility in terms of injections and surgical interventions; its partial immune-privileged properties that limit immune responses towards the antigenicity of the transgene and the viral vectors; and the presence of a tight blood-retinal barrier that can help to prevent unintentional spreading or contamination of the neighboring tissues as well as to the general circulation. In addition, direct corneal delivery, either topically or via injection, can be achieved only in the affected eye in the case of unilateral conditions. The advancement in imaging technologies, such as pentacam and adaptive optics, further facilitates quantitative and qualitative evaluation of corneal changes after gene therapy. However, the wide heterogeneity in disease-causing genes in hereditary corneal disease requires knowledge and identification of the specific causative gene in each patient in order to consider gene therapy as an intervention. However, many of the pathways affected in the tissues as part of their pathology can be common. Therefore, there is a need to design differential strategies according to the causative genes, mutation, and inheritance pattern for gene therapy of various hereditary corneal dystrophies. The success of gene therapy for the treatment of any disease depends on the efficiency by which the therapeutic transgene is delivered to the target cell type. There are currently two main approaches in terms of vectors for delivering genetic material: viral and non-viral vectors.
3.1. Viral Vectors
Significant progress in understanding the molecular mechanism of diverse corneal diseases has led to the development of gene therapies in animal models. Gene therapy in animal models has demonstrated restoration of the biomechanical stability of the cornea by regulating the key proteins that are deficient in various corneal dystrophies, thus establishing a scientific basis for application in human subjects. Most studies on gene therapy of diverse corneal dystrophies have used viral vectors, including adeno-associated virus (AAV), lentivirus, and adenovirus, due to their unique ability to transduce a wide tropism of living cells with minimal resistance. The mechanisms by which the viral vectors deliver the therapeutic gene and translate the therapeutic protein into the host cells is discussed in this section.
3.1.1. Adeno-Associated Virus (AAV)
AAV is a small (25-nm), non-pathogenic, nonenveloped virus that packages a linear single-stranded DNA genome of ~4.7 kb containing two genes that produce 4 Rep and 3 Cap proteins for replication and capsid proteins, respectively (Figure 2). The Rep gene encodes for four different proteins: rep40, rep52, rep68, and rep78. Rep68 and rep78 play an essential role in viral genome integration, replication, and transcriptional regulation of AAV gene expression, whereas rep 40 and rep 52 proteins are involved in viral genome encapsulation. The cap gene encodes three viral capsid proteins, VP1 (90 kDa), VP2 (72 kDa), and VP3 (60 kDa) (Figure 2), which are arranged in a 1:1:10 ratio and form an icosahedral symmetry. AAV belongs to the family Parvoviridae and is placed in the genus Dependovirus [80], as AAV requires a helper virus, such as adenovirus, to establish a productive infection cycle. However, recombinant AAV vectors used for gene therapy do not carry any of its native Rep/Cap genes or any helper genes, underlining its safety profile for therapeutic applications. Additionally, these vectors can transduce and express in both mitotic and post-mitotic cells.
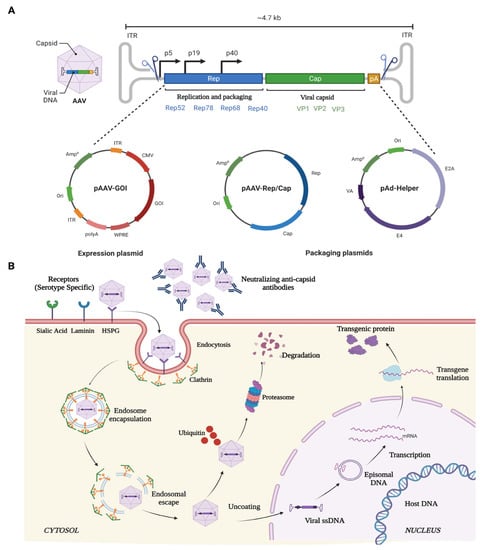
Figure 2. Schematic illustration of (A) AAV genome, and (B) mechanism of entry, infection, and therapeutic gene production in host cell.
The ability of the various rAAV serotypes to transduce a wide variety of ocular structures is due to its ability to bind primary cell surface receptors such as heparin sulphate proteoglycan (HSPGs) [81]. HSPGs are expressed in most of the cell types and uses integrin αvβ5 or fibroblast growth factor receptor (FGFR) as coreceptors for its internalization and endocytosis [82][83]. Once endocytosed, the viral particles are released from the endosome at a low pH [84][85]. The released SS-DNA of AAV is converted to ds-DNA by either annealing to another complementary strand of AAV or via the host cell DNA replication machinery. Subsequently, the viral genome remains as an episome and, with the help of host cell machinery, undergoes transcription and translation to produce the deficient therapeutic protein in the target cells over extended durations (Figure 2).
The ability of the various rAAV serotypes to transduce ocular structures at the anterior segment of the eye has been extensively documented using vectors encoding marker proteins; therefore, it has become evident that a combination of serotypes, route of administration, and the choice of regulatory elements such as promotors allows the selective tropism of desired cell types. Recent studies have also shown that the corneal stroma is an ideal target for AAV-mediated gene therapy where the quiescent stromal keratocytes can receive the vectors and express the therapeutic proteins over long durations. In the context of corneal gene therapy, the first reported gene therapy was mediated by the AAV2 serotype, which demonstrated successful transgene delivery into a rabbit cornea in vivo [86]. The natural occurrence of the AAV2 serotypes may cause its pre-exposure in humans. This exposure leads to the production of neutralizing antibodies against the serotype. Thus, when a therapeutic gene packaged into a AAV2 serotype is delivered to the patient there is a chance of eliciting a humoral immune response against it which in turn may diminish the efficacy and safety of viruses carrying the therapeutic gene. The AAV5 serotype has been found to be the most divergent serotype [87], with a several-fold more efficient transduction efficiency than AAV2 [88]. In vivo gene therapy for corneal scarring using topical administration of the AAV5 serotype in rabbit models has shown a significant decrease in corneal haze and fibrosis, without any reports of any immunogenic or toxic immune response against it [89][90]. In the domain of AAV vectors, AAV8 [91] and AAV9 [92] were considered the most efficient in corneal keratocyte transduction. Therefore, a chimeric AAV capsid was generated where an AAV8 capsid scaffold was engrafted with the AAV9 galactose binding domain. On evaluating the potency of infection of the chimeric AAV8/9 capsid following an intrastromal injection of the vectors into the human donor eyeballs, efficient widespread transduction of the transgene was observed. The AAV8/9 chimeric capsids were found to have greater and more efficient transduction efficiency when compared to either parent serotype at similar vector doses [93]. Therefore, a direct gene augmentation strategy using AAV vectors offers a viable option to cure diseases for which no treatment options exist.
3.1.2. Lentivirus (LV)
Lentiviruses are single-stranded enveloped RNA viruses and are members of the retroviridae family. A mature LV is 100 nm in diameter and has a cylindrical core structure. LV transduction takes place through association with specific cell surface receptors. After binding to the receptor, the viral membrane fuses with the host cell membrane [94] and injects the nucleoprotein complex into the cell. After its entry, the viral RNA is reverse-transcribed into ds-DNA by the reverse transcriptase enzyme associated with the nucleoprotein complex. The ds-DNA enters the nucleus through the nuclear pore complex and is integrated into the host genome with the help of the viral integrase [95] (Figure 3). The cons of using LVs as a vector for corneal gene therapy are that it possesses high immune cell infiltration post-infection, random integration potential [96][97], and a consequent risk of insertional mutagenesis/teratoma formation. Due to these crucial shortcomings, the use of LVs still requires immense evaluation and identification of preferential integration sites in the host genome before being applicable to humans for therapies.
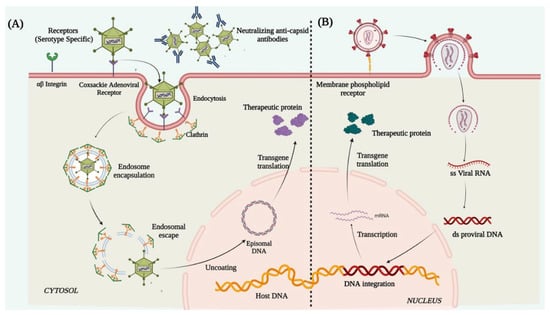
Figure 3. Schematic illustration representing mechanism of action of (A) adenovirus and (B) lentivirus.
3.1.3. Adenovirus (Ad)
Adenoviruses are double-stranded enveloped DNA viruses and are members of the Adenoviridae family. More than 50 known human adenoviral serotypes are present in nature, of which serotypes 2 and 5 are the most widely used in gene therapy. Ad vectors can transduce both dividing and non-dividing cells. It can harbor a therapeutic gene up to 30Kb in size and deliver it to the target tissue. Their inability to integrate into the host genome reduces the chances of insertional mutagenesis. In adenoviral vector preparations, high titers of pure viruses can be easily obtained from a single viral prep, thus a small volume of vector injection into the cornea is enough for a high level of transduction. The Ad vectors deliver the therapeutic gene in the host cell through receptor-mediated endocytosis [98], by binding to the cell surface coxsackie adenovirus receptor, αvβ3 integrin, and clathrin-coated pits. Once internalized, the viral genome containing the therapeutic gene is released into the cytoplasm which further enters the nucleus through the nuclear pore complex and remains distinct from the host genome and is expressed as episomes to produce the therapeutic protein (Figure 3). However, the shortcomings were the short-lived expression of the delivered transgene, primarily due to the rapid histone association and silencing of the Ad genome. This necessitates repeated vector injection, which showed more toxic responses to the cells than the original exposure [99]. The Ad capsid structures (the pentons and heptons) are potent targets for rapid immune recognition causing Ad mediated gene therapy to typically induce significant host immune response [100].
3.2. Non-Viral Vectors
The delivery of the therapeutic gene into the cornea can also be achieved using a non-viral origin vector mediated gene therapy. This includes the use of liposomes, compacted nanoparticles, electroporation, particle gun bombardment, and many other methods. The advantage of non-viral vectors is low immunogenicity, ease of manipulating their chemical properties to suit DNA delivery, the possibility of large-scale production, and transferring large vectors without any immune reactions. However, non-viral vectors may present obstacles such as inefficient cellular membrane transport, intracellular/extracellular degradation, and lack of long-term gene expression [101].
3.2.1. Electroporation
Electroporation is a technique that uses short and intense electric pulses to create pores or reversibly permeabilize the cell membrane in order to deliver the naked circular DNA carrying the therapeutic gene into the target cells (Figure 4). This physical method of non-viral mediated gene therapy has been widely used for successfully delivering plasmid DNA into the corneal endothelium and stromal keratocytes [102][103]. The optimal field strength of 100–200 V/cm was found to be suitable for the transfer of naked plasmid DNA without any corneal damage or inflammation. Various in vivo experiments on different animal models for stromal keratitis and corneal endothelium wound healing experiments showed a 1000-fold increase in gene uptake in the cornea in comparison to injection of DNA alone, leading to inhibition of the disease phenotype [20][104]. One of the main drawbacks when using electroporation is that the permeabilization as a result of the electric pulse becomes irreversible due to the heat generated during the process [105].
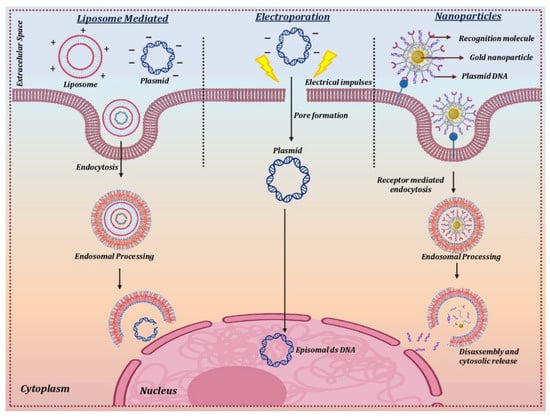
Figure 4. Delivery mechanism for non-viral vectors.
3.2.2. Nanoparticles
Nanoparticles are ultrafine particles that range between 1 and 100 nm in size and are widely used in nanomedicine due to their (a) small size, (b) ability to deliver the transgene into the intracellular compartment of the cells, (c) high surface area to volume ratio, (d) capability to deliver a larger payload, and (e) negligible toxic damage to the cell membrane. The use of nanoparticles is highly suitable for the delivery of transgene for the treatment of eye-related diseases as they can permeabilize across various ocular barriers including the cornea, sclera, conjunctiva, and, in some cases, the blood-retinal barriers. Nanoparticles can harbor multiple cargo types, including DNA, peptides, antibodies, molecular sensors, and drugs, in the desired cellular layers of the eye. Nanoparticles are mainly classified as metallic, polymeric, and hybrid nanoparticles. Polymeric nanoparticles that are usually prepared from albumin, chitosan, and polyethyleneimine (PEI) have been found to be more efficient in delivering the transgene into rodent corneas in vivo without any significant side effects [106][107][108]. In vivo delivery of transgene using hybrid nanoparticles in rabbit cornea to treat corneal fibrosis has demonstrated an efficient cargo of transgene in the target cells with significant inhibition of fibrosis and no visible toxic effects [36].
The direct transfer of the therapeutic gene to the corneal cells in vivo has been achieved using cationic lipids [109]. The positively charged cationic lipids bind to negatively charged DNA molecules to form a lipid-DNA complex that has a high affinity for the cell membrane. The lipid-DNA complex is then endocytosed into an endocytic vesicle followed by its trafficking and release from the endosomal compartment. The nuclear uptake of the DNA takes place through the nuclear pore complex, which then forms an episome to express the therapeutic protein using the host cell machinery [110] (Figure 4).
3.3. Gene Therapy Strategies for Precise and Targeted Therapeutics
3.3.1. Gene Augmentation
Gene augmentation therapy is a straightforward approach in which the transfer of the functional therapeutic gene into the affected cells takes place to restore the expression of an inadequately functioning gene [111][112] (Figure 5). This approach is mainly applicable to recessive genetic diseases. For the augmentation strategy to be effective, the augmented transgene must synthesize physiological and adequate levels of the normal protein and the tissue status and disease stage at which treatment is attempted should not be terminal. To minimize off-target transgene expression that may have a pathogenic effect within the cells, it is important to focus on the appropriate regulation of the transgene expression for augmentation-based approaches. To address appropriate transgene regulation, cell or tissue-specific promoter sequences can be designed to modulate the expression of the therapeutic transgene. For example, in response to inflammation, NFκB responsive promoter sequences have been used to drive the transgene expression in immune organs [113]. A commonly used approach in gene augmentation therapy for secretory protein is to target the delivery of the therapeutic gene to a distant tissue/cell and not the affected cell type or tissue; this will help in the production of the same deficient therapeutic secretory protein to rescue the disease phenotype. This approach can be adopted when (i) transduction of affected cells with the therapeutic gene may not be able to produce a physiologically relevant amount of secreted transgene protein; (ii) the affected site of gene delivery has been rendered sensitive to any further damage that can be caused during the gene delivery procedure; and (iii) the site of gene delivery is highly exposed to the host immune system which can lead to a transgene-directed immune response. Such an approach in corneal gene therapy has not yet been listed in the literature.
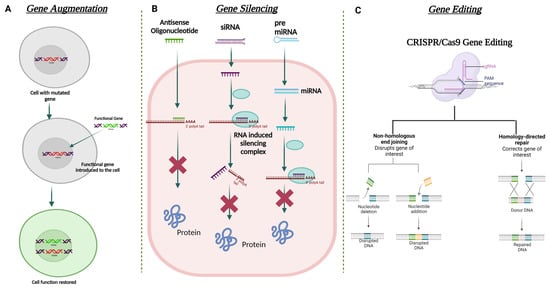
Figure 5. Schematic illustration for the different strategies used in corneal gene therapy. (A) Gene augmentation. (B) Gene silencing using ASO, siRNA, and miRNA. (C) Gene editing using CRISPR Cas9 approach.
Gene augmentation therapy for corneal diseases has been conducted in various animal models in vivo to rescue the disease phenotype. There are numerous studies that have shown that gene augmentation using viral and non-viral vectors can help to significantly improve the disease pathology in murine, ovine, and canine models.
3.3.2. Gene Editing
In recent times, gene editing has become one of the most utilized approaches in the field of gene therapy. This approach is able to target both autosomal dominant and recessively inherited corneal dystrophies that lead to loss of function due to mutation of the target gene. The strategy aims to restore/correct the normal functioning of the mutated gene by adding, removing, or altering the genome at the site of the mutation. One of the most specific and advanced genome editing technologies to date that has been used for gene correction in the anterior segment of the eye is Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins, termed the CRISPR-Cas system [114]. Meganucleases, Zinc finger nucleases (ZFNs) [115], and transcription activator-like effector nucleases (TALENs) are also used in this approach. CRISPR-Cas-based genome editing has been found to be more favorable than the other mentioned nucleases due to its relatively simple design for targeting the mutated gene [116]. On the other hand, significant modifications in the DNA-binding protein domains are required to target with ZFNs and TALENs.
The mechanism of CRISPR-Cas 9 involves the binding of Cas9 nuclease at the genomic locus that is complementary to the guide RNA; this guide RNA creates a double-stranded break, which is repaired via two mechanisms: (i) error-prone non-homologous end joining (NHEJ) or (ii) homologous directed repair (HDR) (Figure 5). NHEJ can create insertions or deletions in the mutated gene, resulting in the formation of a premature stop codon. This can be used to knock out the expression of the truncated protein. HDR can help incorporate specific alterations into the mutated gene provided by the repair template. All studied Cas9 enzymes require a protospacer adjacent motif (PAM) next to the target site for target DNA recognition. Some recent studies have also shown the ability of certain divergent CRISPR-Cas9 enzymes that can recognize and cleave single-stranded DNA (ssDNA) using an RNA-guided, PAM-independent recognition mechanism [117]. Intravenous delivery of the CRISPR-Cas9 system cannot be used in the eye due to the blood-ocular barrier [118]. AAV as a vector has been widely used for the delivery of CRISPR-Cas9 construct into the target tissue/cell. However, the biggest shortcoming of using AAV vectors is their carrying capacity. They are often too small to accommodate the full genome editing system. To address this issue, a dual-AAV vector design has been implemented in which the Cas9 nuclease and the single-guide RNA (sgRNA) cassettes have been packaged in two separate vectors and delivered to the target tissue, which showed highly efficient genome editing in the hepatocyte cells [119]. The CRISPR-Cas 9 system has been used to treat corneal dystrophy in animal models. In one study, selective disruption of the mutated copy of the KRT12 gene in a humanized MECD mouse model was achieved by targeting the protospacer adjacent motif (PAM) sequence that was generated by the missense mutation in the KRT12 gene [120].
3.3.3. Gene Silencing
Gene augmentation therapy is likely to be ineffective in rescuing the disease phenotype in autosomal dominant diseases. For augmentation therapy to work effectively, the suppression of the mutated gene expression must be addressed first. Silencing the mutated gene can be mediated via delivering a small double-stranded non-coding RNA construct that is designed to act via RNA interference (RNAi) [121]. RNAi promotes post-transcriptional gene silencing by enzymatic degradation of the complementary RNA species. This silencing is mediated by a large multi-component RNA/protein complex called the RNA-induced silencing complex (RISC) [122] (Figure 5). siRNAs (small interfering RNA) have been tested in various corneal dystrophies, including MECD, wound healing, and neovascularization [123][124][125].
In the context of mRNA-based gene therapy, the most successful approach is the use of antisense oligonucleotides (AONs). AONs are short single-stranded DNA or RNA that interacts with the complementary mRNA to block the translation by altering the pre-mRNA splicing [126]. The first AON approved by the FDA was for the treatment of cytomegalovirus retinitis: fomivirsen (Vitravene). In the cornea, the first phase clinical trial was conducted to explore the efficacy of a topically administered AON (Aganirsen) that targets the insulin substrate-1 receptor to block corneal neovascularization in keratitis patients. The AON administration was found effective and thus reduced the need for corneal transplantation [127]. GS-101 AON was also found to be a potent anti-angiogenic compound that helped in the significant regression of corneal neovascularization [128].
Despite gaining success, the strategy of gene silencing fails to completely silence the target gene expression. For the RNAi pathway, the major drawbacks, such as off-the-mark effects and extended toxicity of the small RNA molecules, must be considered and studied carefully for its successful application in clinical settings.
3.3.4. Dual Vectors
Limitations of using rAAV-based gene therapy correctional strategies include its limited packaging size for genes larger than 4.7 kb such as PD-L1, which is widely used in corneal graft rejection studies. Several strategies have been designed to take advantage of the head-tail concatamerization of the AAV genome (Figure 6). One strategy is to use the overlapping sequence approach. In this method, efficient reconstitution and transgene expression rely on the use of two separate (dual) AAV vectors, each one carrying half of a large gene. Upon coinfection of the target cell from both vectors, the two halves will be reconstituted due to the canonical ability of AAV genomes to concatamerize via intermolecular recombination. In 2007, Ghosh et al. [129] developed a hybrid dual vector strategy to expand the packaging capacity in the rAAV system, the trans-splicing vectors, and showed effective, whole-body transduction using a mouse model of Duchenne Muscular Dystrophy. This trans-splicing strategy employs the use of a splice donor at the 3′ end of one-half of the target gene in one vector and a splice acceptor at the 5′ end of the other half of the transgene. This allows for efficient reconstitution of the mRNA transcripts by the head-to-tail concatamerization process. The third dual AAV approach (hybrid) is a combination of the two previous approaches; it is based on the addition of a highly recombinogenic exogenous sequence (recombinogenic region) to the trans-splicing vectors in order to increase their recombination efficiency. In 2011, Ghosh et al. [130] reported the use of minimized bridging sequences from the highly recombinogenic alkaline phosphatase (~227–247 bp) to circumvent the available space for packaging the transgene. In this approach, the authors demonstrated the complete reconstitution of the B-Galactosidase gene (LacZ) when packaged separately and co-infected to check expression. Expression of the reconstituted LacZ was proven to be better than the original hybrid strategy developed in suitable cell cultures and animal models.
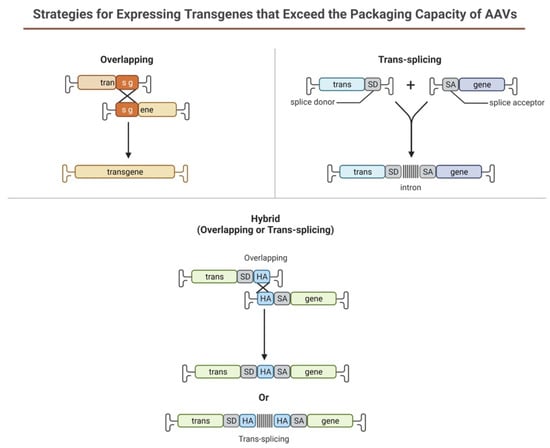
Figure 6. Schematic illustration representing dual AAV approach for large genes.
This entry is adapted from the peer-reviewed paper 10.3390/cells12091280
References
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221.
- Prado, D.A.; Acosta-Acero, M.; Maldonado, R.S. Gene therapy beyond luxturna: A new horizon of the treatment for inherited retinal disease. Curr. Opin. Ophthalmol. 2020, 31, 147–154.
- Mashhour, B.; Couton, D.; Perricaudet, M.; Briand, P. In vivo adenovirus-mediated gene transfer into ocular tissues. Gene Ther. 1994, 1, 122–126.
- Constantin, C. Corneal dystrophies: Pathophysiological, genetic, clinical, and therapeutic considerations. Rom. J. Ophthalmol. 2021, 65, 104–108.
- Lisch, W.; Weiss, J.S. Early and late clinical landmarks of corneal dystrophies. Exp. Eye Res. 2020, 198, 108139.
- Loukovitis, E.; Sfakianakis, K.; Syrmakesi, P.; Tsotridou, E.; Orfanidou, M.; Bakaloudi, D.R.; Stoila, M.; Kozei, A.; Koronis, S.; Zachariadis, Z.; et al. Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities. Ophthalmol. Ther. 2018, 7, 263–292.
- Sonoda, S.; Tachibana, K.; Uchino, E.; Okubo, A.; Yamamoto, M.; Sakoda, K.; Hisatomi, T.; Sonoda, K.H.; Negishi, Y.; Izumi, Y.; et al. Gene transfer to corneal epithelium and keratocytes mediated by ultrasound with microbubbles. Investig. Ophthalmol. Vis. Sci. 2006, 47, 558–564.
- Stechschulte, S.U.; Joussen, A.M.; von Recum, H.A.; Poulaki, V.; Moromizato, Y.; Yuan, J.; D’Amato, R.J.; Kuo, C.; Adamis, A.P. Rapid ocular angiogenic control via naked DNA delivery to cornea. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1975–1979.
- Huang, Y.-X.; Li, Q.-H. An active artificial cornea with the function of inducing new corneal tissue generation in vivo—A new approach to corneal tissue engineering. Biomed. Mater. 2007, 2, S121.
- Jhanji, V.; Mehta, J.S.; Sharma, N.; Sharma, B.; Vajpayee, R.B. Targeted corneal transplantation. Curr. Opin. Ophthalmol. 2012, 23, 324–329.
- Chamberlain, W.D. Femtosecond laser-assisted deep anterior lamellar keratoplasty. Curr. Opin. Ophthalmol. 2019, 30, 256–263.
- Luengo-Gimeno, F.; Tan, D.T.; Mehta, J.S. Evolution of deep anterior lamellar keratoplasty (DALK). Ocul. Surf. 2011, 9, 98–110.
- Klausner, E.A.; Peer, D.; Chapman, R.L.; Multack, R.F.; Andurkar, S.V. Corneal gene therapy. J. Control. Release 2007, 124, 107–133.
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173.
- Fuest, M.; Yam, G.H.; Peh, G.S.; Mehta, J.S. Advances in corneal cell therapy. Regen. Med. 2016, 11, 601–615.
- Mobaraki, M.; Abbasi, R.; Omidian Vandchali, S.; Ghaffari, M.; Moztarzadeh, F.; Mozafari, M. Corneal Repair and Regeneration: Current Concepts and Future Directions. Front. Bioeng. Biotechnol. 2019, 7, 135.
- Mohan, R.R.; Rodier, J.T.; Sharma, A. Corneal gene therapy: Basic science and translational perspective. Ocul. Surf. 2013, 11, 150–164.
- Torrecilla, J.; Del Pozo-Rodriguez, A.; Vicente-Pascual, M.; Solinis, M.A.; Rodriguez-Gascon, A. Targeting corneal inflammation by gene therapy: Emerging strategies for keratitis. Exp. Eye Res. 2018, 176, 130–140.
- Sakamoto, T.; Oshima, Y.; Nakagawa, K.; Ishibashi, T.; Inomata, H.; Sueishi, K. Target gene transfer of tissue plasminogen activator to cornea by electric pulse inhibits intracameral fibrin formation and corneal cloudiness. Hum. Gene Ther. 1999, 10, 2551–2557.
- Oshima, Y.; Sakamoto, T.; Yamanaka, I.; Nishi, T.; Ishibashi, T.; Inomata, H. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther. 1998, 5, 1347–1354.
- Williams, K.A.; Jessup, C.F.; Coster, D.J. Gene therapy approaches to prolonging corneal allograft survival. Expert Opin. Biol. Ther. 2004, 4, 1059–1071.
- Lakshminarayanan, R.; Chaurasia, S.S.; Anandalakshmi, V.; Chai, S.M.; Murugan, E.; Vithana, E.N.; Beuerman, R.W.; Mehta, J.S. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul. Surf. 2014, 12, 234–251.
- Jonsson, F.; Bystrom, B.; Davidson, A.E.; Backman, L.J.; Kellgren, T.G.; Tuft, S.J.; Koskela, T.; Ryden, P.; Sandgren, O.; Danielson, P.; et al. Mutations in collagen, type XVII, alpha 1 (COL17A1) cause epithelial recurrent erosion dystrophy (ERED). Hum. Mutat. 2015, 36, 463–473.
- Irvine, A.D.; Corden, L.D.; Swensson, O.; Swensson, B.; Moore, J.E.; Frazer, D.G.; Smith, F.J.; Knowlton, R.G.; Christophers, E.; Rochels, R.; et al. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann’s corneal dystrophy. Nat. Genet. 1997, 16, 184–187.
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100.
- Hassan, H.; Thaung, C.; Ebenezer, N.D.; Larkin, G.; Hardcastle, A.J.; Tuft, S.J. Severe Meesmann’s epithelial corneal dystrophy phenotype due to a missense mutation in the helix-initiation motif of keratin 12. Eye 2013, 27, 367–373.
- Nishida, K.; Honma, Y.; Dota, A.; Kawasaki, S.; Adachi, W.; Nakamura, T.; Quantock, A.J.; Hosotani, H.; Yamamoto, S.; Okada, M.; et al. Isolation and chromosomal localization of a cornea-specific human keratin 12 gene and detection of four mutations in Meesmann corneal epithelial dystrophy. Am. J. Hum. Genet. 1997, 61, 1268–1275.
- Szaflik, J.P.; Oldak, M.; Maksym, R.B.; Kaminska, A.; Pollak, A.; Udziela, M.; Ploski, R.; Szaflik, J. Genetics of Meesmann corneal dystrophy: A novel mutation in the keratin 3 gene in an asymptomatic family suggests genotype-phenotype correlation. Mol. Vis. 2008, 14, 1713–1718.
- Wieben, E.D.; Aleff, R.A.; Tang, X.; Butz, M.L.; Kalari, K.R.; Highsmith, E.W.; Jen, J.; Vasmatzis, G.; Patel, S.V.; Maguire, L.J.; et al. Trinucleotide Repeat Expansion in the Transcription Factor 4 (TCF4) Gene Leads to Widespread mRNA Splicing Changes in Fuchs’ Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 343–352.
- Jordan, T.; Hanson, I.; Zaletayev, D.; Hodgson, S.; Prosser, J.; Seawright, A.; Hastie, N.; van Heyningen, V. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992, 1, 328–332.
- Tomatsu, S.; Montano, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519.
- Garrido, E.; Cormand, B.; Hopwood, J.J.; Chabas, A.; Grinberg, D.; Vilageliu, L. Maroteaux-Lamy syndrome: Functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol. Genet. Metab. 2008, 94, 305–312.
- Massague, J.; Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000, 19, 1745–1754.
- Kim, W.J.; Mohan, R.R.; Mohan, R.R.; Wilson, S.E. Effect of PDGF, IL-1alpha, and BMP2/4 on corneal fibroblast chemotaxis: Expression of the platelet-derived growth factor system in the cornea. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1364–1372.
- Dudley, A.T.; Lyons, K.M.; Robertson, E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995, 9, 2795–2807.
- Tandon, A.; Sharma, A.; Rodier, J.T.; Klibanov, A.M.; Rieger, F.G.; Mohan, R.R. BMP7 gene transfer via gold nanoparticles into stroma inhibits corneal fibrosis in vivo. PLoS ONE 2013, 8, e66434.
- Saika, S.; Ikeda, K.; Yamanaka, O.; Miyamoto, T.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Nakajima, Y.; Kao, W.W.; et al. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am. J. Pathol. 2005, 166, 1405–1418.
- Wilson, S.E.; Chen, L.; Mohan, R.R.; Liang, Q.; Liu, J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp. Eye Res. 1999, 68, 377–397.
- Li, J.F.; Duan, H.F.; Wu, C.T.; Zhang, D.J.; Deng, Y.; Yin, H.L.; Han, B.; Gong, H.C.; Wang, H.W.; Wang, Y.L. HGF accelerates wound healing by promoting the dedifferentiation of epidermal cells through beta1-integrin/ILK pathway. Biomed. Res. Int. 2013, 2013, 470418.
- Chang, J.H.; Gabison, E.E.; Kato, T.; Azar, D.T. Corneal neovascularization. Curr. Opin. Ophthalmol. 2001, 12, 242–249.
- Kvanta, A.; Sarman, S.; Fagerholm, P.; Seregard, S.; Steen, B. Expression of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor (VEGF) in inflammation-associated corneal neovascularization. Exp. Eye Res. 2000, 70, 419–428.
- Klettner, A.; Roider, J. Treating age-related macular degeneration—Interaction of VEGF-antagonists with their target. Mini Rev. Med. Chem. 2009, 9, 1127–1135.
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371.
- Stevenson, W.; Cheng, S.F.; Dastjerdi, M.H.; Ferrari, G.; Dana, R. Corneal neovascularization and the utility of topical VEGF inhibition: Ranibizumab (Lucentis) vs bevacizumab (Avastin). Ocul. Surf. 2012, 10, 67–83.
- Yoon, K.C.; Ahn, K.Y.; Lee, J.H.; Chun, B.J.; Park, S.W.; Seo, M.S.; Park, Y.G.; Kim, K.K. Lipid-mediated delivery of brain-specific angiogenesis inhibitor 1 gene reduces corneal neovascularization in an in vivo rabbit model. Gene Ther. 2005, 12, 617–624.
- Cho, Y.K.; Uehara, H.; Young, J.R.; Tyagi, P.; Kompella, U.B.; Zhang, X.; Luo, L.; Singh, N.; Archer, B.; Ambati, B.K. Flt23k nanoparticles offer additive benefit in graft survival and anti-angiogenic effects when combined with triamcinolone. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2328–2336.
- Lai, C.M.; Shen, W.Y.; Brankov, M.; Lai, Y.K.; Barnett, N.L.; Lee, S.Y.; Yeo, I.Y.; Mathur, R.; Ho, J.E.; Pineda, P.; et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol. Ther. 2005, 12, 659–668.
- Li, Z.; He, T.; Du, K.; Xing, Y.Q.; Run, Y.M.; Yan, Y.; Shen, Y. Inhibition of oxygen-induced ischemic retinal neovascularization with adenoviral 15-lipoxygenase-1 gene transfer via up-regulation of PPAR-gamma and down-regulation of VEGFR-2 expression. PLoS ONE 2014, 9, e85824.
- Qazi, Y.; Stagg, B.; Singh, N.; Singh, S.; Zhang, X.; Luo, L.; Simonis, J.; Kompella, U.B.; Ambati, B.K. Nanoparticle-mediated delivery of shRNA.VEGF-a plasmids regresses corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2837–2844.
- Torrecilla, J.; Gomez-Aguado, I.; Vicente-Pascual, M.; Del Pozo-Rodriguez, A.; Solinis, M.A.; Rodriguez-Gascon, A. MMP-9 Downregulation with Lipid Nanoparticles for Inhibiting Corneal Neovascularization by Gene Silencing. Nanomaterials 2019, 9, 631.
- Yu, H.; Chen, L.; Jiang, J. Administration of pigment epithelium-derived factor delivered by adeno-associated virus inhibits blood-retinal barrier breakdown in diabetic rats. Mol. Vis. 2010, 16, 2384–2394.
- Coster, D.J.; Williams, K.A. The impact of corneal allograft rejection on the long-term outcome of corneal transplantation. Am. J. Ophthalmol. 2005, 140, 1112–1122.
- Parker, D.G.; Brereton, H.M.; Coster, D.J.; Williams, K.A. The potential of viral vector-mediated gene transfer to prolong corneal allograft survival. Curr. Gene Ther. 2009, 9, 33–44.
- Beutelspacher, S.C.; Pillai, R.; Watson, M.P.; Tan, P.H.; Tsang, J.; McClure, M.O.; George, A.J.; Larkin, D.F. Function of indoleamine 2,3-dioxygenase in corneal allograft rejection and prolongation of allograft survival by over-expression. Eur. J. Immunol. 2006, 36, 690–700.
- Gong, N.; Pleyer, U.; Volk, H.D.; Ritter, T. Effects of local and systemic viral interleukin-10 gene transfer on corneal allograft survival. Gene Ther. 2007, 14, 484–490.
- Gong, N.; Pleyer, U.; Yang, J.; Vogt, K.; Hill, M.; Anegon, I.; Volk, H.D.; Ritter, T. Influence of local and systemic CTLA4Ig gene transfer on corneal allograft survival. J. Gene Med. 2006, 8, 459–467.
- Pillai, R.G.; Beutelspacher, S.C.; Larkin, D.F.; George, A.J. Expression of the chemokine antagonist vMIP II using a non-viral vector can prolong corneal allograft survival. Transplantation 2008, 85, 1640–1647.
- Williams, K.A.; Coster, D.J. Gene therapy for diseases of the cornea—A review. Clin. Exp. Ophthalmol. 2010, 38, 93–103.
- Murthy, R.C.; McFarland, T.J.; Yoken, J.; Chen, S.; Barone, C.; Burke, D.; Zhang, Y.; Appukuttan, B.; Stout, J.T. Corneal transduction to inhibit angiogenesis and graft failure. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1837–1842.
- Comer, R.M.; King, W.J.; Ardjomand, N.; Theoharis, S.; George, A.J.; Larkin, D.F. Effect of administration of CTLA4-Ig as protein or cDNA on corneal allograft survival. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1095–1103.
- Tang, X.L.; Sun, J.F.; Wang, X.Y.; Du, L.L.; Liu, P. Blocking neuropilin-2 enhances corneal allograft survival by selectively inhibiting lymphangiogenesis on vascularized beds. Mol. Vis. 2010, 16, 2354–2361.
- Cho, Y.K.; Zhang, X.; Uehara, H.; Young, J.R.; Archer, B.; Ambati, B. Vascular Endothelial Growth Factor Receptor 1 morpholino increases graft survival in a murine penetrating keratoplasty model. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8458–8471.
- Swierkowska, J.; Gajecka, M. Genetic factors influencing the reduction of central corneal thickness in disorders affecting the eye. Ophthalmic Genet. 2017, 38, 501–510.
- Dudakova, L.; Sasaki, T.; Liskova, P.; Palos, M.; Jirsova, K. The presence of lysyl oxidase-like enzymes in human control and keratoconic corneas. Histol. Histopathol. 2016, 31, 63–71.
- Shetty, R.; Sathyanarayanamoorthy, A.; Ramachandra, R.A.; Arora, V.; Ghosh, A.; Srivatsa, P.R.; Pahuja, N.; Nuijts, R.M.; Sinha-Roy, A.; Mohan, R.R.; et al. Attenuation of lysyl oxidase and collagen gene expression in keratoconus patient corneal epithelium corresponds to disease severity. Mol. Vis. 2015, 21, 12–25.
- Smith, V.A.; Matthews, F.J.; Majid, M.A.; Cook, S.D. Keratoconus: Matrix metalloproteinase-2 activation and TIMP modulation. Biochim. Biophys. Acta 2006, 1762, 431–439.
- Shetty, R.; Ghosh, A.; Lim, R.R.; Subramani, M.; Mihir, K.; Reshma, A.R.; Ranganath, A.; Nagaraj, S.; Nuijts, R.M.; Beuerman, R.; et al. Elevated expression of matrix metalloproteinase-9 and inflammatory cytokines in keratoconus patients is inhibited by cyclosporine A. Investig. Ophthalmol. Vis. Sci. 2015, 56, 738–750.
- Shetty, R.; D’Souza, S.; Khamar, P.; Ghosh, A.; Nuijts, R.; Sethu, S. Biochemical Markers and Alterations in Keratoconus. Asia Pac. J. Ophthalmol. 2020, 9, 533–540.
- Brancati, F.; Valente, E.M.; Sarkozy, A.; Feher, J.; Castori, M.; Del Duca, P.; Mingarelli, R.; Pizzuti, A.; Dallapiccola, B. A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J. Med. Genet. 2004, 41, 188–192.
- Burdon, K.P.; Coster, D.J.; Charlesworth, J.C.; Mills, R.A.; Laurie, K.J.; Giunta, C.; Hewitt, A.W.; Latimer, P.; Craig, J.E. Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum. Genet. 2008, 124, 379–386.
- Hameed, A.; Khaliq, S.; Ismail, M.; Anwar, K.; Ebenezer, N.D.; Jordan, T.; Mehdi, S.Q.; Payne, A.M.; Bhattacharya, S.S. A novel locus for Leber congenital amaurosis (LCA4) with anterior keratoconus mapping to chromosome 17p13. Investig. Ophthalmol. Vis. Sci. 2000, 41, 629–633.
- Hutchings, H.; Ginisty, H.; Le Gallo, M.; Levy, D.; Stoesser, F.; Rouland, J.F.; Arne, J.L.; Lalaux, M.H.; Calvas, P.; Roth, M.P.; et al. Identification of a new locus for isolated familial keratoconus at 2p24. J. Med. Genet. 2005, 42, 88–94.
- Wheeler, J.; Hauser, M.A.; Afshari, N.A.; Allingham, R.R.; Liu, Y. The Genetics of Keratoconus: A Review. Reprod. Syst. Sex Disord. 2012.
- Czugala, M.; Karolak, J.A.; Nowak, D.M.; Polakowski, P.; Pitarque, J.; Molinari, A.; Rydzanicz, M.; Bejjani, B.A.; Yue, B.Y.; Szaflik, J.P.; et al. Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur. J. Hum. Genet. 2012, 20, 389–397.
- Droitcourt, C.; Touboul, D.; Ged, C.; Ezzedine, K.; Cario-Andre, M.; de Verneuil, H.; Colin, J.; Taieb, A. A prospective study of filaggrin null mutations in keratoconus patients with or without atopic disorders. Dermatology 2011, 222, 336–341.
- Guan, T.; Liu, C.; Ma, Z.; Ding, S. The point mutation and polymorphism in keratoconus candidate gene TGFBI in Chinese population. Gene 2012, 503, 137–139.
- Heon, E.; Greenberg, A.; Kopp, K.K.; Rootman, D.; Vincent, A.L.; Billingsley, G.; Priston, M.; Dorval, K.M.; Chow, R.L.; McInnes, R.R.; et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum. Mol. Genet. 2002, 11, 1029–1036.
- Lechner, J.; Dash, D.P.; Muszynska, D.; Hosseini, M.; Segev, F.; George, S.; Frazer, D.G.; Moore, J.E.; Kaye, S.B.; Young, T.; et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3215–3223.
- Udar, N.; Atilano, S.R.; Brown, D.J.; Holguin, B.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1: A candidate gene for keratoconus. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3345–3351.
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756.
- O’Donnell, J.; Taylor, K.A.; Chapman, M.S. Adeno-associated virus-2 and its primary cellular receptor--Cryo-EM structure of a heparin complex. Virology 2009, 385, 434–443.
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77.
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. AlphaVbeta5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82.
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 2000, 74, 2777–2785.
- Douar, A.M.; Poulard, K.; Stockholm, D.; Danos, O. Intracellular trafficking of adeno-associated virus vectors: Routing to the late endosomal compartment and proteasome degradation. J. Virol. 2001, 75, 1824–1833.
- Mohan, R.R.; Schultz, G.S.; Hong, J.W.; Mohan, R.R.; Wilson, S.E. Gene transfer into rabbit keratocytes using AAV and lipid-mediated plasmid DNA vectors with a lamellar flap for stromal access. Exp. Eye Res. 2003, 76, 373–383.
- Chiorini, J.A.; Kim, F.; Yang, L.; Kotin, R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999, 73, 1309–1319.
- Zabner, J.; Seiler, M.; Walters, R.; Kotin, R.M.; Fulgeras, W.; Davidson, B.L.; Chiorini, J.A. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J. Virol. 2000, 74, 3852–3858.
- Gupta, S.; Rodier, J.T.; Sharma, A.; Giuliano, E.A.; Sinha, P.R.; Hesemann, N.P.; Ghosh, A.; Mohan, R.R. Targeted AAV5-Smad7 gene therapy inhibits corneal scarring in vivo. PLoS ONE 2017, 12, e0172928.
- Mohan, R.R.; Tandon, A.; Sharma, A.; Cowden, J.W.; Tovey, J.C. Significant inhibition of corneal scarring in vivo with tissue-selective, targeted AAV5 decorin gene therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4833–4841.
- Hippert, C.; Ibanes, S.; Serratrice, N.; Court, F.; Malecaze, F.; Kremer, E.J.; Kalatzis, V. Corneal transduction by intra-stromal injection of AAV vectors in vivo in the mouse and ex vivo in human explants. PLoS ONE 2012, 7, e35318.
- Sharma, A.; Tovey, J.C.; Ghosh, A.; Mohan, R.R. AAV serotype influences gene transfer in corneal stroma in vivo. Exp. Eye Res. 2010, 91, 440–448.
- Vance, M.; Llanga, T.; Bennett, W.; Woodard, K.; Murlidharan, G.; Chungfat, N.; Asokan, A.; Gilger, B.; Kurtzberg, J.; Samulski, R.J.; et al. AAV Gene Therapy for MPS1-associated Corneal Blindness. Sci. Rep. 2016, 6, 22131.
- Maddon, P.J.; McDougal, J.S.; Clapham, P.R.; Dalgleish, A.G.; Jamal, S.; Weiss, R.A.; Axel, R. HIV infection does not require endocytosis of its receptor, CD4. Cell 1988, 54, 865–874.
- Gonda, M.A.; Wong-Staal, F.; Gallo, R.C.; Clements, J.E.; Narayan, O.; Gilden, R.V. Sequence homology and morphologic similarity of HTLV-III and visna virus, a pathogenic lentivirus. Science 1985, 227, 173–177.
- Mohan, R.R.; Martin, L.M.; Sinha, N.R. Novel insights into gene therapy in the cornea. Exp. Eye Res. 2021, 202, 108361.
- Mohan, R.R.; Tovey, J.C.; Sharma, A.; Schultz, G.S.; Cowden, J.W.; Tandon, A. Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS ONE 2011, 6, e26432.
- Petrie, N.C.; Yao, F.; Eriksson, E. Gene therapy in wound healing. Surg. Clin. N. Am. 2003, 83, 597–616.
- Borras, T.; Gabelt, B.T.; Klintworth, G.K.; Peterson, J.C.; Kaufman, P.L. Non-invasive observation of repeated adenoviral GFP gene delivery to the anterior segment of the monkey eye in vivo. J. Gene Med. 2001, 3, 437–449.
- Fisher, K.D.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001, 8, 341–348.
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128.
- Chang, Y.K.; Hwang, J.S.; Chung, T.Y.; Shin, Y.J. SOX2 Activation Using CRISPR/dCas9 Promotes Wound Healing in Corneal Endothelial Cells. Stem Cells 2018, 36, 1851–1862.
- Zhou, R.; Dean, D.A. Gene transfer of interleukin 10 to the murine cornea using electroporation. Exp. Biol. Med. (Maywood) 2007, 232, 362–369.
- Oshima, Y.; Sakamoto, T.; Hisatomi, T.; Tsutsumi, C.; Sassa, Y.; Ishibashi, T.; Inomata, H. Targeted gene transfer to corneal stroma in vivo by electric pulses. Exp. Eye Res. 2002, 74, 191–198.
- Rosazza, C.; Meglic, S.H.; Zumbusch, A.; Rols, M.P.; Miklavcic, D. Gene Electrotransfer: A Mechanistic Perspective. Curr. Gene Ther. 2016, 16, 98–129.
- de la Fuente, M.; Seijo, B.; Alonso, M.J. Bioadhesive hyaluronan-chitosan nanoparticles can transport genes across the ocular mucosa and transfect ocular tissue. Gene Ther. 2008, 15, 668–676.
- Jain, G.K.; Pathan, S.A.; Akhter, S.; Jayabalan, N.; Talegaonkar, S.; Khar, R.K.; Ahmad, F.J. Microscopic and spectroscopic evaluation of novel PLGA-chitosan Nanoplexes as an ocular delivery system. Colloids Surf. B Biointerfaces 2011, 82, 397–403.
- Nagarwal, R.C.; Singh, P.N.; Kant, S.; Maiti, P.; Pandit, J.K. Chitosan nanoparticles of 5-fluorouracil for ophthalmic delivery: Characterization, in-vitro and in-vivo study. Chem. Pharm. Bull. 2011, 59, 272–278.
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417.
- Farhood, H.; Serbina, N.; Huang, L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim. Biophys. Acta 1995, 1235, 289–295.
- Hu, M.L.; Edwards, T.L.; O’Hare, F.; Hickey, D.G.; Wang, J.H.; Liu, Z.; Ayton, L.N. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin. Exp. Optom. 2021, 104, 444–454.
- Lee, J.H.; Wang, J.H.; Chen, J.; Li, F.; Edwards, T.L.; Hewitt, A.W.; Liu, G.S. Gene therapy for visual loss: Opportunities and concerns. Prog. Retin. Eye Res. 2019, 68, 31–53.
- Chtarto, A.; Bockstael, O.; Gebara, E.; Vermoesen, K.; Melas, C.; Pythoud, C.; Levivier, M.; De Witte, O.; Luthi-Carter, R.; Clinkers, R.; et al. An adeno-associated virus-based intracellular sensor of pathological nuclear factor-kappaB activation for disease-inducible gene transfer. PLoS ONE 2013, 8, e53156.
- Burnight, E.R.; Giacalone, J.C.; Cooke, J.A.; Thompson, J.R.; Bohrer, L.R.; Chirco, K.R.; Drack, A.V.; Fingert, J.H.; Worthington, K.S.; Wiley, L.A.; et al. CRISPR-Cas9 genome engineering: Treating inherited retinal degeneration. Prog. Retin. Eye Res. 2018, 65, 28–49.
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555.
- Raikwar, S.P.; Raikwar, A.S.; Chaurasia, S.S.; Mohan, R.R. Gene editing for corneal disease management. World J. Transl. Med. 2016, 5, 1–13.
- Ma, E.; Harrington, L.B.; O’Connell, M.R.; Zhou, K.; Doudna, J.A. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 2015, 60, 398–407.
- Yu, W.; Wu, Z. Ocular delivery of CRISPR/Cas genome editing components for treatment of eye diseases. Adv. Drug Deliv. Rev. 2021, 168, 181–195.
- Yang, Y.; Wang, L.; Bell, P.; McMenamin, D.; He, Z.; White, J.; Yu, H.; Xu, C.; Morizono, H.; Musunuru, K.; et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016, 34, 334–338.
- Courtney, D.G.; Moore, J.E.; Atkinson, S.D.; Maurizi, E.; Allen, E.H.; Pedrioli, D.M.; McLean, W.H.; Nesbit, M.A.; Moore, C.B. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2016, 23, 108–112.
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498.
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296.
- Courtney, D.G.; Atkinson, S.D.; Allen, E.H.; Moore, J.E.; Walsh, C.P.; Pedrioli, D.M.; MacEwen, C.J.; Pellegrini, G.; Maurizi, E.; Serafini, C.; et al. siRNA silencing of the mutant keratin 12 allele in corneal limbal epithelial cells grown from patients with Meesmann’s epithelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3352–3360.
- Liu, Q.; Wu, K.; Qiu, X.; Yang, Y.; Lin, X.; Yu, M. siRNA silencing of gene expression in trabecular meshwork: RhoA siRNA reduces IOP in mice. Curr. Mol. Med. 2012, 12, 1015–1027.
- Supe, S.; Upadhya, A.; Singh, K. Role of small interfering RNA (siRNA) in targeting ocular neovascularization: A review. Exp. Eye Res. 2021, 202, 108329.
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406.
- Cursiefen, C.; Viaud, E.; Bock, F.; Geudelin, B.; Ferry, A.; Kadlecova, P.; Levy, M.; Al Mahmood, S.; Colin, S.; Thorin, E.; et al. Aganirsen antisense oligonucleotide eye drops inhibit keratitis-induced corneal neovascularization and reduce need for transplantation: The I-CAN study. Ophthalmology 2014, 121, 1683–1692.
- Cursiefen, C.; Bock, F.; Horn, F.K.; Kruse, F.E.; Seitz, B.; Borderie, V.; Fruh, B.; Thiel, M.A.; Wilhelm, F.; Geudelin, B.; et al. GS-101 antisense oligonucleotide eye drops inhibit corneal neovascularization: Interim results of a randomized phase II trial. Ophthalmology 2009, 116, 1630–1637.
- Ghosh, A.; Duan, D. Expanding adeno-associated viral vector capacity: A tale of two vectors. Biotechnol. Genet. Eng. Rev. 2007, 24, 165–177.
- Ghosh, A.; Yue, Y.; Duan, D. Efficient transgene reconstitution with hybrid dual AAV vectors carrying the minimized bridging sequences. Hum. Gene Ther. 2011, 22, 77–83.
This entry is offline, you can click here to edit this entry!