Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Tumor-associated macrophages (TAMs) are the earliest infiltrating cells in PanIN lesions and continue to rise throughout cancer progression. TAMs are the most abundant immune cells in the stroma and are the key drivers shaping the immunosuppressive landscape. There are various mechanisms employed to polarize macrophages to tumor-supporting subtypes which subsequently unleash the plethora of neoplastic characteristics, including promoting malignant cell proliferation, augmenting angiogenesis, metastasis, acquiring pleiotropic pancreatic cancer stem-like cells, supporting chemoresistance.
- angiogenesis
- chemoresistance
- epithelial–mesenchymal transition
- macrophage
- tumor-associated macrophage
- metastasis
- pancreatic ductal adenocarcinoma
- PDAC
- tumor microenvironment
- immunosuppression
1. Introduction
1.1. Introduction of Pancreatic Cancer
Pancreatic carcinoma is the deadliest malignancy afflicting the exocrine digestive organ. This cancer is well known for lacking screening tools and having early metastatic spread, followed by chemoresistance, leading to limited treatment strategies and poor prognostic outcomes [1][2]. As such, it took 466,003 lives across 185 countries in 2020 and is presently the seventh leading cause of deaths from cancers in both genders [3]. Trends forecasted through 2040 predict that pancreatic cancer will become the second-most-leading cause of cancer-related death in the United States [4], and approximately 355,317 new cases will occur globally [5]. Among them, nearly 95% of pancreatic cancer incidences are pancreatic ductal adenocarcinoma (PDAC) [6]. Approximately 80% of pancreatic cancer patients present with advanced-to-late stages of nonresectable and disseminated disease [7]. The two most common first-line chemotherapeutic regimens include blends of 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) and gemcitabine (GEM) plus Nab-Paclitaxel [8]. However, therapeutic intervention scarcely improves overall prognosis, and the 5-year survival rate remains disappointing [9][10].
PDAC develops sporadically but is largely due to the acquisition of constitutively active mutant Kirsten rat sarcoma (KRAS) derived from the most frequent driver mutations: G12R, G12V, and G12D, which comprise approximately 90% of occurrence [11]. Among them, G12D accounts for about 40% of incidents [11][12]. Initial progression to pancreatic cancer embarks from the cells harboring KRAS mutations engaging in networking with proinflammatory cytokines [13][14][15]. For instance, in response to oncogenic mutant KRAS, interleukin (IL)-6 induces the expression and activation of signal transducer and activator of transcription 3 (STAT3) [15][16][17][18]. Accordingly, persistent STAT3 signaling was demonstrated to play a pivotal role in mutant KRAS-induced pancreatic tumorigenesis [19], and demonstrated that Janus kinase (JAK)–STAT3 axis activation correlates with a poor outcome in PDAC patients following surgical resections [20]. Moreover, oncogenic mutant KRAS unleashes a plethora of signaling cascades, including rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK), and phosphoinositol 3-kinase (PI3K)/protein kinase B (AKT) pathways in various malignant entities including pancreatic cancer [11][21][22][23]. RAF/MEK/ERK is the first well-known Ras effector in cancers. GTP-bound KRAS interacts with and triggers RAF, which further induces the phosphorylation and activation of MEK1 and MEK2. This scenario subsequently enhances ERK1 and ERK2 serine/threonine kinases activities. Activated ERK1/2 then phosphorylates over 200 targets, many of which are transcription factors controlling cell proliferation [24][25]. Mounting evidence demonstrates the critical role of PI3K being a regulator for embarking oncogenic KRAS-driven carcinogenesis, largely by governing cell survival and proliferation [26][27]. Another independent study utilizing a genetically engineered mouse model containing mutant Kras elucidates a similar finding that the PI3K pathway can augment PDAC through the activation of STAT3 and nuclear factor kappa B (NF-κB) signaling [28].
The first histological alteration occurring in PDAC pathogenesis is the transdifferentiation of acinar cells into duct-like cells, named acinar-to-ductal metaplasia (ADM) [29][30]. The molecular causes underlying dysregulated ADM were recently elucidated to be associated with a loss of AT-rich interactive domain containing protein 1A (ARID1A) [31], followed by interaction between PAF1 (RNA polymerase II-associated factor 1) and YAP1 (yes activated protein-1) [32]. For ADM, infiltrating macrophages secrete inflammatory cytokines including regulated on activation normal T cell expressed and secreted (RANTES) [33] and tumor necrosis factor-alpha (TNF-α). Together, they lead to the activation of NF-κB signaling and expression of matrix metalloproteinases (MMPs) [33][34]. In response to chronic inflammation, acinar pancreatic cells adopt ADM [29] and then develop precancerous lesions, which are not only frequently observed in pancreatitis [35], but also develop into pancreatic intraepithelial neoplasia (PanIN) following the acquisition of oncogenic mutations such as KRAS [29]. Both ADM and PanIN constitute crucial aberrations in PDAC and persist throughout tumor development [34][36]. During this neoplastic progression, macrophage depletion not only blocks the progression of ADM to PanIN, but also lightens PDAC burden in mice [34][37], underscoring the imperative role played by these immune cells.
Although oncogenic Kras mutation in mouse PDAC is critical for cancer initiation, constitutively activated mutant KRAS alone is insufficient for tumor onset; rather, it requires partner mutations such as the P53 tumor suppressor gene, as well as cytokines produced by different cell types within the tumor mass [38]. A genetically engineered mouse model combining both mutations, LSL-KrasG12D; Trp53flox/flox; Pdx-1-Cre (KPC), has been established as a clinically relevant PDAC model that recapitulates many key features of human PDAC with a robust inflammatory response [39] and elevated immunosuppressive features [40].
1.2. Introduction of Tumor Microenvironment and Immune Evasion
Marked by extensive fibrosis and inflammation, PDAC’s tumor microenvironment (TME) consists of fibroblasts, immune cells, endothelial cells, and an acellular extracellular matrix (ECM) that contains various growth factors, chemokines, and cytokines [41]. Within the TME, cancer cells interplay with nearby stroma and acellular constituents that synergistically controls malignant traits and therapeutic outcomes [42][43]. Fibroblastic stroma can hinder drug entry by safeguarding tumor cells from therapeutic insults [44], and then advancing tumor progression characterized by invasion, angiogenesis, metastasis, and chemoresistance [45]. PDAC is initially featured with chronic inflammation triggered by immune aberrations [46]. Then, oncogenic mutant KRAS augments inflammation and launches an immunosuppressive TME that subsequently plays a pivotal role in cancer progression [47][48][49][50].
In general, immune responses are modulated by a plethora of checkpoint regulators that act as “security brakes” and establish a “do not eat me” cue when inflammation reactions shall be ended from prior infections, or autoimmunity shall be circumvented by enhancing self-tolerance. Cancers exploit various immune checkpoint modulators, attempting to evade tumoricidal responses, favor immune tolerance, and escape recognition and clearance by immune surveillance cells [51]. Therapeutic agents abolishing such functions are recognized as immune checkpoint blockades (ICBs) that have been proven to improve clinical outcomes [52]. Yet, PDAC remains largely embraced by an immunosuppressive TME with limited infiltration of tumoricidal immune cells, thereby resulting in a poor response to ICBs [53][54]. The TME attracts several immunosuppressive cell types that circumvent the surveillance normally conducted by cytotoxic cluster of differentiated (CD)8+ T lymphocytes and by dendritic cells (DC) [53].
Within the TME of PDAC, infiltration of tumoricidal CD8+ T lymphocytes is rare. Accordingly, a few well-known ICBs attempting to revive T lymphocytes to date have manifested disappointing efficacy [55]. Instead, the tumor bed is infiltrated with largely protumorigenic immune-suppressive cells including myeloid-derived suppressor cells (MDSC), regulatory T cells (Treg), and tumor-associated macrophages (TAM) [47][53]. TAMs are the earliest infiltrating cells in PanIN lesions and continue to rise throughout cancer progression [56]. Macrophages in PDAC are derived from blends of circulating monocytes and phagocytes that reside in the pancreas. Moreover, TAMs are the most abundant immune cells in the stroma and are the key drivers shaping the immunosuppressive landscape [57]. TAMs enhance tumor immune evasion, mainly by enhancing tumor fibrosis and excluding tumoricidal T lymphocytes [58]. TAM infiltration not only correlates with lymph node metastasis and poor prognosis [59], but also plays multifaceted roles in the carcinogenesis of PDAC [60].
As a vital innate immune population for maintaining body homeostasis and warding off foreign particles or pathogens, macrophages can regularly sense their microenvironment, display high plasticity, and execute diverse functions adapted to different environmental contexts. Depending on the inflammatory cues, macrophages can develop two distinct subtypes, these being either classically activated M1 or alternatively activated M2 subpopulations [61]. M1 macrophages are proinflammatory and tumoricidal, whereas M2 macrophages are anti-inflammatory, protumorigenic, and immunosuppressive [61][62]. Furthermore, fully polarized macrophages can depolarize and transform reciprocally in response to environmental triggers [63]. The M1 subtype commonly produces higher levels of IL-1, IL-6, IL-12, IL-23, TNFα, chemokine C-X-C motif ligand (CXCL)9, CXCL10, and inducible nitric oxide synthase (iNOS) [64]. Conversely, the M2-type commonly expresses higher levels of IL-10, transforming growth factor-β1 (TGF-β1), and arginase 1 (ARG1) [65][66][67][68][69][70]. M2 is the most abundant immunosuppressive subpopulation representing approximately 85% of TAMs [53][57][71][72]. Infiltration and the abundance of M2 is not only a malignant hallmark but also correlates with poor prognosis [73][74]. Yang et al. demonstrated that targeting proliferating F4/80+ macrophages by the pharmacological inhibitor, clodronate liposomes, fostered CD8+ T cell infiltration and promoted their spatial redistribution, thereby enhancing antitumor immunity [75]. Furthermore, closer proximity of M2 macrophages to the tumor core strongly correlates with poor disease-free survival [69], highlighting the clinical impact of M2 macrophages on molding a cancer-promoting landscape [61][71][76].
Macrophages exist on a spectrum of polarization states between the M1 and M2 phenotypic extremes and exhibit functional plasticity within the TME [77]. The early stages of tumor lesions initially have a high abundance of M1 macrophages that are later polarized to the M2 population as PDAC progresses [78]. Preclinical and clinical trials have been completed, or are still ongoing, attempting to target TAMs and treat various cancer types including pancreatic cancer (e.g., NCT03662412, NCT03184870, and NCT01921699) [79]. Although M2 macrophages are still under substantive studies, this aims to extrapolate PDAC-fostered M2 macrophages, delineate TME-orchestrated mechanisms responsible for M2 polarization, and then discern how the M2 population synergizes cancer cells and TME factors to convey multifaceted impacts on PDAC.
2. Factors Modulate Polarization of TAM
2.1. Factors Released from Malignant Cells or Cancer-Associated Fibroblasts (CAFs)
Crosstalk between neoplastic cells and infiltrating macrophages in the tumor milieu governs PDAC carcinogenesis. TAMs are in close contact with cancer-secreted factors and thereby are polarized towards the M2 phenotype [37][80][81]. Intriguingly, oncogenic mutant KRAS can recruit TAMs and then promote carcinogenesis [80]. Mutant KRAS not only releases growth factors but also regulates glucose metabolism in PDAC [82]. Accordingly, lactate and granulocyte-macrophage colony-stimulating factor (GM-CSF) are known to be profoundly released from cancer cells expressing oncogenic mutant KRAS [82][83] (Figure 1). This aberration is mediated through the PI3K/AKT signaling cascade that partly enhances macrophage polarization [84]. Moreover, regenerating gene family member 4 (REG4) released from PDAC cancer cells [85][86] can promote macrophage polarization to M2, as well as orchestrate the TME to favor cancer growth and metastasis [87] (Figure 1). Consequently, high numbers of M2-polarized TAMs correlate with an increased incidence of lymph node metastasis [87]. The underlying molecular mechanism accounting for this scenario was deemed to be mediated through the epidermal growth factor receptor (EGFR)/AKT/cAMP-response element binding protein (CREB) signaling pathway [87]. A further study elucidated that the overexpression of recombinant REG4 enhanced the expression of IL-10, CD163, and many other M2 signature genes in TAMs [87]. Additionally, the secretion of IL-10 can be upregulated by insulin-like growth factor binding protein 2 (IGFBP2) released from cancer cells following STAT3 activation [88] (Figure 1). IGFBP2 favors M2 macrophages and exacerbates an immunosuppressive TME by increasing Treg infiltration and inhibiting antitumor T cell immunity in a mouse model [88]. Hence, blocking the IGFBP2 axis constitutes a promising treatment protocol through which TAM polarization can be attenuated and a tumoricidal state of the TME can be revived [88]. Together, multiple networks maneuver TAM polarization toward an M2 state.
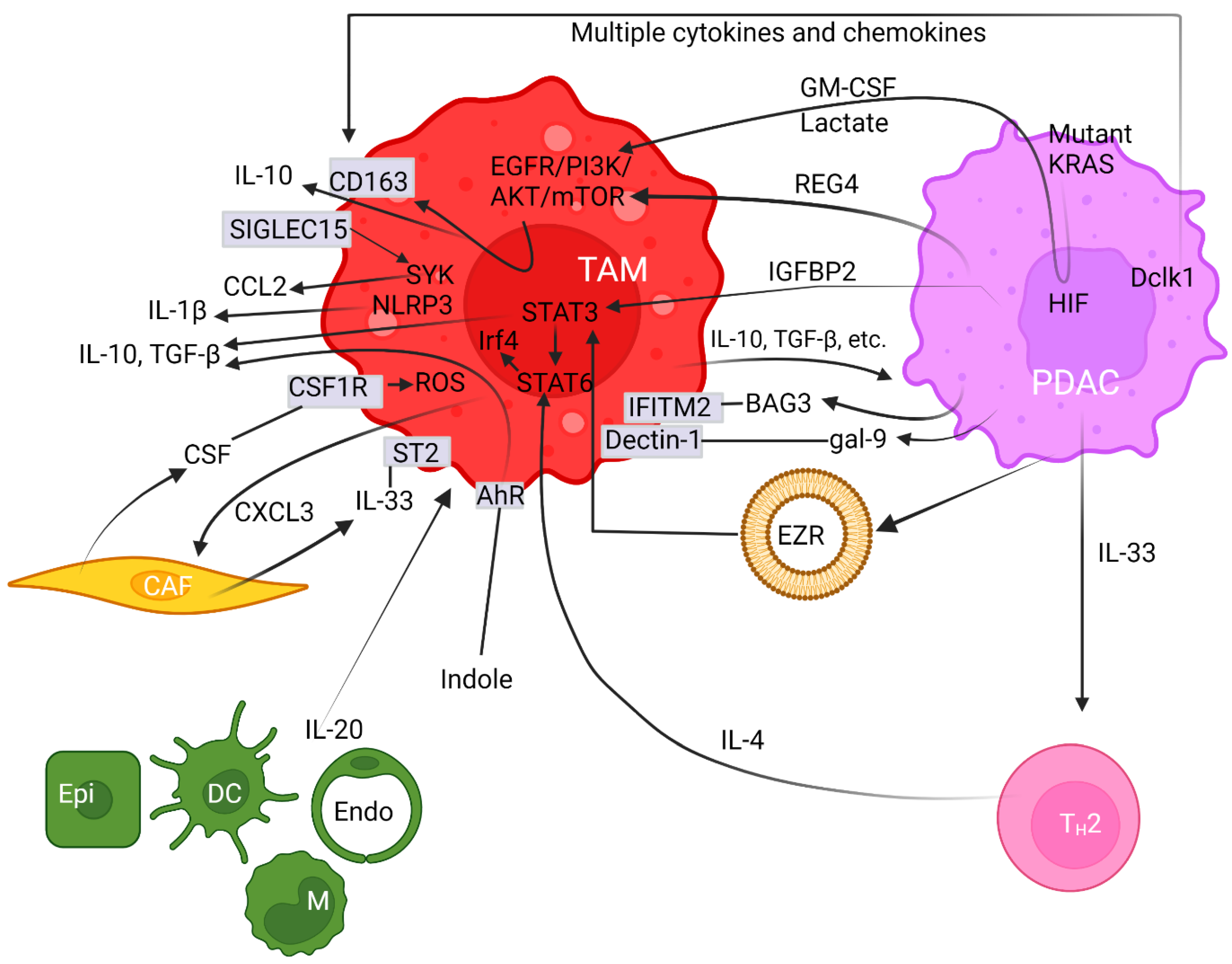
Figure 1. Pancreatic ductal adenocarcinoma cells synergize with the tumor microenvironment to provoke polarization of M2 macrophages. Arrows with pointed or with blocked ends indicate activation or inhibition between regulators, respectively, while a fading effect at the start of arrows represents secretion of modulators. Plain straight lines depict interaction between ligands and receptors. Cell surface proteins are noted in rectangular boxes on cell membranes. The circular lipid bilayer depicts an extracellular vesicle. Abbreviations used include aryl hydrocarbon receptor (AhR), protein kinases B (AKT), Bcl-2-associated athanogene 3 (BAG3), cancer-associated fibroblast (CAF), CC-chemokine ligand (CCL), cluster of differentiated (CD), CXC chemokine ligand (CXCL), colony-stimulating factor (CSF) and receptor (CSF1R), dendritic cell (DC), double cortin-like kinase 1 (Dclk1), endothelial cell (Endo), epidermal growth factor receptor (EGFR), epithelial cell (Epi), ezrin (EZR), galectin (gal), granulocyte-macrophage colony-stimulating factor (GM-CSF), hypoxia-inducible factor (HIF), interleukin (IL), insulin-like growth factor binding protein 2 (IGFBP2), interferon-induced transmembrane protein 2 (IFITM-2), interferon regulatory factor 4 (Irf4), Kirsten rat sarcoma (KRAS), mammalian target of rapamycin (mTOR), microRNA (miR), monocyte (M), nucleotide-binding and leucine-rich repeat receptor containing pyrin domain 3 (NLRP3), pancreatic ductal adenocarcinoma (PDAC), phosphatidylinositol 3-kinase (PI3K), reactive oxygen species (ROS), regenerating gene family member 4 (REG4), sialic-acid-binding immunoglobulin-like lectin 15 (SIGLEC15), signal transducer and activator of transcription (STAT), spleen tyrosine kinase (SYK), suppression of tumorigenicity 2 (ST2), tumor-associated macrophage (TAM), transforming growth factor β (TGF-β), and T helper-2 (TH2).
Double cortin-like kinase 1 (Dclk1) is overexpressed in the cancer cores and PanIN lesions, based off various pancreatic cancer models [89] (Figure 1). By releasing various chemokines and cytokines, the elevated Dclk1-isoform 2 resulted in the polarization towards the M2 phenotype (Figure 1). This aberration is demarcated by a high abundance of M2 macrophages and low occupancy of CD8+ T cell infiltration with weakened tumoricidal activities [90]. These M2 macrophages enhance cell migration, invasion, and self-renewal, along with increased expression of Snail and Slug, both of which are indicatives of cancer stem-like cells [90][91]. Moreover, galectin-9 (gal-9), a member of the P-galactoside-binding family of lectins, was found to be highly expressed in both mouse and human PDAC. The binding of gal-9 to its receptor, Dectin-1, a crucial innate immune regulator expressed on the surface of macrophages, polarizes macrophages to the M2 phenotype (Figure 1). Disruption of the gal-9/dectin-1 interaction reverts immunosuppression, enhances cytotoxic T lymphocytes recruitment, downregulates Tregs, impedes tumor growth, and achieves improved therapeutic efficacy [92][93][94]. Moreover, Ezrin (EZR) expression is upregulated in PDAC and is associated with tumor progression [95]. Chang et al. demonstrated that extracellular vesicles (EVs)-capsulated EZR is strikingly correlated with poor survival in PDAC patients [96]. Molecular investigations further discerned that overexpressed EZR regulates STAT3 activation [97] that further synergizes with STAT6 to augment the polarization of TAMs towards the M2 phenotypes [98] (Figure 1). Consistently, Su et al. reported miR-155 and miR-125b2 as being the key regulator encapsulated in the PDAC cell-line-derived EV that exploits a dose-dependent effect on macrophage plasticity [99].
On the other hand, CAFs release colony-stimulating factor (CSF) and induce M2 polarization through binding to receptor CSF1R within the PDAC milieu, and then enhance reactive oxygen species (ROS) production in monocytes [100] (Figure 1). The importance of ROS activation on M2 polarization was illustrated by the evidence that ROS ablation abrogates this effect [101]. Anti-CSF1R therapy favors the M1-like subpopulation in vivo, thereby exerting a powerful antitumor effect on glioma neoplasm [102]. Furthermore, stromal fibroblasts are the predominant cell types for producing IL-33 that mainly targets its receptor, known as suppression of tumorigenicity 2 (ST2), on TAMs and induces the polarization of M2 [103][104]. Upon activation, IL-33-polarized TAMs subsequently release CXCL3 to further amplify CAFs. Together, this interactive axis constitutes a paracrine and positive feedback loop amplifying both CAF and TAM cell types [105] (Figure 1).
2.2. Factors Produced from Stromal Immune Cells
Abundantly in PDAC, oncogenic mutant Kras can activate the downstream PI3K/AKT/mammalian target of the rapamycin (mTOR) signaling pathway [106]. Consequently, the aberrant activation of this cascade conveys tumor initiation, cancer progression, and metastatic spread, followed by emerging chemoresistance [107]. This signaling axis can be effectively abrogated by urolithin A (Uro A) [108]. The treatment of PDAC cells with Uro A not only inhibited the growth of tumor xenografts and improved the overall survival (OS) of Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice, but also reprogrammed the tumor microenvironment by attenuating infiltrated immunosuppressive cells such as TAMs, MDSCs, and Tregs [108].
Oncogenic mutant KrasG12D elevates IL-33 expression in PDAC cells, which recruits and activates TH2 cells. Then, TH2 cells stimulate tumor growth by secreting protumorigenic cytokines such as IL-4 that exerts major impacts on neighboring innate immune cells (Figure 1). Studies on animal models unveiled that IL-4-initiated signaling in macrophages can be further orchestrated by Stat6, which in turn regulates interferon regulatory factor 4 (Irf4) that acts as an important transcription factor and harnesses M2 polarization [109] (Figure 1). Conversely, Irf4 deficiency impeded the expression of M2-associated signature genes [110]. In a syngeneic model of PDAC, the inhibition of Irf4 using the immunomodulatory agent pomalidomide resulted in a shift of macrophages towards the M1 population and fosters an immune surveillance antitumor environment along with an improved infiltration of cytotoxic T lymphocytes and enhanced immune responses [111].
Proinflammatory cytokine IL-20 is a member of the IL-10 family and is expressed predominantly by epithelial cells, monocytes, dendritic cells, and endothelial cells in the TME (Figure 1). IL-20 was demonstrated to promote M2 polarization, and elevated IL-20 levels in PDAC tumor tissue correlate with poor overall survival [112]. Inhibiting IL-20 using an antagonistic antibody, 7E, reshapes the TME toward scenarios unfavorable for malignancies in multiple aspects including diminished M2 macrophage infiltration, lightened fibrosis, inhibited tumor growth, and reduced expression of the immunosuppressive regulator PD-L1 on tumor cells [112].
TAMs remain the primary cell type molding the immune landscape [75][113], partly fortified by a self-amplifying mechanism. Sialic-acid-binding immunoglobulin-like lectin 15 (SIGLEC15) is upregulated in M2 macrophages and could directly enact immunosuppressive function via binding α-2,3 sialic acid [114]. Stimulation of the extracellular domain of SIGLEC15 promotes the tyrosine phosphorylation of DNAX-activating protein of 12 kDa (DAP12) and leads to the activation and recruitment of spleen tyrosine kinase (SYK) [115] (Figure 1). Joshi et al. further revealed an autocrine-positive feedback loop phenomenon by demonstrating that SYK, in conjunction with the PI3K axis, synergizes M2 polarization, which can be abolished by a dual SYK/PI3K inhibitor, SRX3207 [116]. α-2,3 sialic-acid-bound SIGLEC15 enhances the production of C-C motif chemokine ligand (CCL)2, C-X-C motif chemokine (CXCL)2, and CXCL8 in TAMs, which not only exacerbates immune suppression but also accelerates tumor progression in gastric [117], esophageal [118], and bladder carcinomas [119]. Among them, CCL2 facilitates the mobilization of receptor CCR2+ inflammatory monocytes from bone marrow to the tumor bed, where they become immunosuppressive TAMs [120]. Together, SIGLEC15 expression, monocyte mobilization, and M2 polarization form a positive feedback circuit, enabling the recruitment and amplification of TAMs [114]. In this regard, a clinical trial in patients with nonmetastatic PDAC using the orally dosed small-molecule CCR2 inhibitor (CCR2i) PF-04136309, in combination with FOLFIRINOX, demonstrated improved antitumor efficacy (trial number NCT01413022). However, a compensatory influx of CXCR2+ neutrophils resulted in a relapse. Yet, this therapeutic resistance can be circumvented by combinatorial blockades targeting both types of infiltrating myeloid cells. Dual treatments not only promote a robust antitumor effect compared to either inhibitor alone, but also improve the overall response to FOLFIRINOX [121].
On the other hand, CD40, a cell surface receptor belonging to the TNF superfamily, can regulate myeloid cell function and adaptive immunity. Similar to Toll-like receptors (TLRs), the CD40 pathway acts as a linkage between DCs and adaptive immunity in cancer. Ligands of CD40 (CD40L) connect DCs and other immune cells in response to malignancies or pathogenic insults with memory. Yet, agonistic anti-CD40 (αCD40) monoclonal antibodies mimic CD40L in vivo and have been shown to enhance the immunogenicity of cancer vaccines and trigger cancer regressions [122][123][124], including in pancreatic cancer [125]. Interestingly, one of the well-studied αCD40 antibodies, selicrelumab, was taken into clinical evaluation as a novel agent for immune activation and cancer immunotherapy, independent from ICB [126]. CD40 activation by selicrelumab enhanced the polarization of TAMs towards the M1 phenotype, as well as activated the proliferation and infiltration of CD8+ T lymphocytes and DCs [126][127]. Together, this treatment transforms the TME from “cold” to “hot” immunity [126][127]. Surgical samples from patients receiving selicrelumab preoperatively exhibited decreased tumor fibrosis, fewer M2 macrophages, and a greater maturation of intratumoral DCs [127]. It is noteworthy to mention that, clinically, combinatorial treatments using αCD40 antibodies and ICB ameliorate efficacy in patients who are initially refractory to immunotherapies. Accordingly, Winograd et al. developed an effective treatment regimen with αCD40 antibodies and ICB (αPD-1/αCTLA-4) using a genetically engineered KPC mouse model [128]. Such success exemplified that the combination of αCD40/ICB, but not either of αCD40 or ICB alone, results in a prominent decline in tumor burden and gain of immunological memory [128].
2.3. Aberrant Metabolism, Hypoxic TME, and Dysregulated Epigenetics
Indole compounds are evolved from dietary tryptophane upon metabolizations by gut microbials such as lactobacilli. Indoles are the key activators for aryl hydrocarbon receptor (AhR), although tryptophane metabolism by human cells rendered negligible effects. By promoting the polarization of TAMs to M2, elevated AhR expression has been recognized as a central driver of TAM function in responding to multiple cues to promote an immune-suppressive state of the TME [129] (Figure 1). Molecular studies delineate that high expression of AhR inhibits IFNγ expression in CD8+ T cells [129], while it enhances the expression of immunosuppressive IL-10 [130], TGF-β, and Arg1 [131][132] (Figure 1). The aforementioned data on animal models coincide with clinical facts in which patients with high AhR expression are strongly correlated with rapid disease progression and increased mortality, along with the immune-suppressive properties associated with TAMs, underscoring the conservation of this regulatory axis in PDAC [129].
PDAC ubiquitously fosters a hypoxic TME. Hypoxia is a condition where the oxygen pressure is below 5–10 mm of mercury, and this phenomenon can empower cancer metastasis [133]. The major mechanism executing cellular responses toward hypoxia is the activation and sustainment of hypoxia-inducible factors (HIFs), mainly HIF1 and HIF2, that activate a set of genes facilitating tumor growth, angiogenesis, and metastasis [134][135]. On the other hand, the endocytosis of cancer, or immune or endothelial cells, can form and release extracellular exosomes [136]. The tumor-derived exosomal miR-301a-3p, for example, not only is released from hypoxic PDAC, but also promotes M2 polarization and ameliorates the PTEN/PI3Kγ pathway, thereby enhancing metastasis in vitro and in vivo [137]. Stimulated by a hypoxic TME, HIF-1α further augments the expression of glycolytic enzymes contributing to maintaining bioenergetic homeostasis during hypoxic stress [138]. In support of this notion, inflammatory cells such as TAMs tend to maneuver metabolism toward glycolysis to meet high energetic demand [139]. Recent studies have unveiled that hypoxia and glycolysis-related gene signatures are concurrently associated with an unfavorable TME and are used to predict a poor prognosis of PDAC patients [140]. Hypoxia and glycolysis pathways are upregulated in the prognostically high-risk cohorts compared to the low-risk counterparts [141][142]. Apart from glucose metabolism, the ablation of HIF2 in CAFs modestly reduces fibrosis and significantly decreases the intratumoral recruitment of M2 macrophages and Treg cells. Similarly, treatment with the clinical HIF2 inhibitor PT2399 abolishes paracrine signaling driven by HIF2, and significantly reduces M2 polarization as well as improves tumor responses to immunotherapy using ICB in PDAC mouse models [143].
GEM treatment favors TAM infiltration into the tumor mass and shifts the stroma to a predominantly M2 phenotype that conveys notorious survival [79], owing to the destruction of gemcitabine by M2-released pyrimidines [144]. Furthermore, paracrine signals from the removal of chemotherapy-generated apoptotic cells can stimulate immune-suppressive controllers in the TME. The phagocytosis of apoptotic cells increases the production of TGF-β1, prostaglandin E2 (PGE2), and platelet-activating factor (PAF), all of which are known to act as anti-inflammatory and immune-suppressive modulators [145].
Dysregulated epigenetic modulators can influence TAM polarization. An epigenomic analysis of TAMs isolated from PDAC tissues revealed the overexpression of CCCTC binding factor (CTCF), an important epigenetic regulator in TAMs. CTCF can enhance M2 polarization and favor the tumor-promoting properties of the TAMs. CTCF-transcribed long noncoding RNA (LncRNA) of prostaglandin-endoperoxide synthase 2 (PTGS2) antisense NF-κB1 complex-mediated expression regulator RNA (PACERR) can orchestrate PTGS2 expression. A novel investigation demarcated that transcribed LncRNA PACERR binds CTCF, forming the CTCF/PACERR complex to recruit the E1A binding protein p300 (EP300), which is one of the histone acetyltransferases. Being an epigenetic regulator, this complex not only enhances chromatin accessibility, but also elevates PTGS2 transcription. Excessively expressed PTGS2 is one of the key activators for polarizing M2 [146].
Moreover, cancer progression and the chemoresistance of PDACs have been associated with elevated histone deacetylases (HDACs) and glycogen synthase kinase 3 beta (GSK3B) activity. Accordingly, treatment by the dual inhibitor, metavert, lowers the abundance of M2 macrophages by more than 50%, although the total number of macrophages are unaffected significantly [147]. These data implicate the molecular cue leading to cancer inhibition by metavert is partially due to the reversion of M2 to the M1 phenotype [147]. Metavert treatment further downregulates procancer cytokines like IL-6 and IL-4, induces cancer cell apoptosis, and attenuates the expression of cancer stem cell markers, as well as impedes cancer growth and metastases [147].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15133507
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- Ungefroren, H.; Konukiewitz, B.; Braun, R.; Wellner, U.F.; Keck, T.; Marquardt, J.U. Elucidation of the Role of SMAD4 in Epithelial-Mesenchymal Plasticity: Does It Help to Better Understand the Consequences of DPC4 Inactivation in the Malignant Progression of Pancreatic Ductal Adenocarcinoma? Cancers 2023, 15, 581.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708.
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27.
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403.
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18.
- Di Costanzo, F.; Di Costanzo, F.; Antonuzzo, L.; Mazza, E.; Giommoni, E. Optimizing First-Line Chemotherapy in Metastatic Pancreatic Cancer: Efficacy of FOLFIRINOX versus Nab-Paclitaxel Plus Gemcitabine. Cancers 2023, 15, 416.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Li, J.; Li, Y.; Chen, C.; Guo, J.; Qiao, M.; Lyu, J. Recent estimates and predictions of 5-year survival rate in patients with pancreatic cancer: A model-based period analysis. Front. Med. 2022, 9, 1049136.
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Shen, H.; Lundy, J.; Strickland, A.H.; Harris, M.; Swan, M.; Desmond, C.; Jenkins, B.J.; Croagh, D. KRAS G12D Mutation Subtype in Pancreatic Ductal Adenocarcinoma: Does It Influence Prognosis or Stage of Disease at Presentation? Cells 2022, 11, 3175.
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302.
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; Lopez-Millan, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 Functions through the Novel REG3beta-JAK2-STAT3 Inflammatory Pathway to Promote the Transition from Chronic Pancreatitis to Pancreatic Cancer. Cancer Res. 2015, 75, 4852–4862.
- Siddiqui, I.; Erreni, M.; Kamal, M.A.; Porta, C.; Marchesi, F.; Pesce, S.; Pasqualini, F.; Schiarea, S.; Chiabrando, C.; Mantovani, A.; et al. Differential role of Interleukin-1 and Interleukin-6 in K-Ras-driven pancreatic carcinoma undergoing mesenchymal transition. Oncoimmunology 2018, 7, e1388485.
- Steele, C.W.; Kaur Gill, N.A.; Jamieson, N.B.; Carter, C.R. Targeting inflammation in pancreatic cancer: Clinical translation. World J. Gastrointest. Oncol. 2016, 8, 380–388.
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Gorgulu, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated with Patient Survival. Gastroenterology 2016, 151, 180–193.e112.
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575.
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029.
- Denley, S.M.; Jamieson, N.B.; McCall, P.; Oien, K.A.; Morton, J.P.; Carter, C.R.; Edwards, J.; McKay, C.J. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 2013, 17, 887–898.
- McCormick, F. K-Ras protein as a drug target. J. Mol. Med. 2016, 94, 253–258.
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070.
- Kim, H.J.; Lee, H.N.; Jeong, M.S.; Jang, S.B. Oncogenic KRAS: Signaling and Drug Resistance. Cancers 2021, 13, 5599.
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121.
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226.
- Castellano, E.; Downward, J. Role of RAS in the regulation of PI 3-kinase. Curr. Top Microbiol. Immunol. 2010, 346, 143–169.
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274.
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schonhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev. 2014, 28, 2621–2635.
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304.
- Parte, S.; Nimmakayala, R.K.; Batra, S.K.; Ponnusamy, M.P. Acinar to ductal cell trans-differentiation: A prelude to dysplasia and pancreatic ductal adenocarcinoma. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188669.
- Zhang, Z.; Wang, X.; Hamdan, F.H.; Likhobabina, A.; Patil, S.; Aperdannier, L.; Sen, M.; Traub, J.; Neesse, A.; Fischer, A.; et al. NFATc1 Is a Central Mediator of EGFR-Induced ARID1A Chromatin Dissociation During Acinar Cell Reprogramming. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1219–1246.
- Nimmakayala, R.K.; Ogunleye, A.O.; Parte, S.; Krishna Kumar, N.; Raut, P.; Varadharaj, V.; Perumal, N.K.; Nallasamy, P.; Rauth, S.; Cox, J.L.; et al. PAF1 cooperates with YAP1 in metaplastic ducts to promote pancreatic cancer. Cell Death Dis. 2022, 13, 839.
- Huang, X.; Li, X.; Ma, Q.; Xu, Q.; Duan, W.; Lei, J.; Zhang, L.; Wu, Z. Chronic alcohol exposure exacerbates inflammation and triggers pancreatic acinar-to-ductal metaplasia through PI3K/Akt/IKK. Int. J. Mol. Med. 2015, 35, 653–663.
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577.
- Song, S.Y.; Gannon, M.; Washington, M.K.; Scoggins, C.R.; Meszoely, I.M.; Goldenring, J.R.; Marino, C.R.; Sandgren, E.P.; Coffey, R.J., Jr.; Wright, C.V.; et al. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 1999, 117, 1416–1426.
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483.
- Liou, G.Y.; Doppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63.
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373.
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14.39.11–14.39.20.
- Yang, J.; Eresen, A.; Shangguan, J.; Ma, Q.; Yaghmai, V.; Zhang, Z. Irreversible electroporation ablation overcomes tumor-associated immunosuppression to improve the efficacy of DC vaccination in a mice model of pancreatic cancer. Oncoimmunology 2021, 10, 1875638.
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311.
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84.
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 296–306.
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49.
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6.
- Jiang, W.; Chen, C.; Huang, L.; Shen, J.; Yang, L. GATA4 Regulates Inflammation-Driven Pancreatic Ductal Adenocarcinoma Progression. Front. Cell Dev. Biol. 2021, 9, 640391.
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205.
- Bansod, S.; Dodhiawala, P.B.; Lim, K.H. Oncogenic KRAS-Induced Feedback Inflammatory Signaling in Pancreatic Cancer: An Overview and New Therapeutic Opportunities. Cancers 2021, 13, 5481.
- Lefler, J.E.; MarElia-Bennett, C.B.; Thies, K.A.; Hildreth, B.E., 3rd; Sharma, S.M.; Pitarresi, J.R.; Han, L.; Everett, C.; Koivisto, C.; Cuitino, M.C.; et al. STAT3 in tumor fibroblasts promotes an immunosuppressive microenvironment in pancreatic cancer. Life Sci. Alliance 2022, 5, e202201460.
- Falcomata, C.; Barthel, S.; Schneider, G.; Rad, R.; Schmidt-Supprian, M.; Saur, D. Context-Specific Determinants of the Immunosuppressive Tumor Microenvironment in Pancreatic Cancer. Cancer Discov. 2023, 13, 278–297.
- Swoboda, A.; Nanda, R. Immune Checkpoint Blockade for Breast Cancer. Cancer Treat. Res. 2018, 173, 155–165.
- Michel, L.L.; von Au, A.; Mavratzas, A.; Smetanay, K.; Schutz, F.; Schneeweiss, A. Immune Checkpoint Blockade in Patients with Triple-Negative Breast Cancer. Target. Oncol. 2020, 15, 415–428.
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14.
- Loch, F.N.; Kamphues, C.; Beyer, K.; Schineis, C.; Rayya, W.; Lauscher, J.C.; Horst, D.; Dragomir, M.P.; Schallenberg, S. The Immune Checkpoint Landscape in Tumor Cells of Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 2160.
- Stromnes, I.M.; Hulbert, A.; Rollins, M.R.; Basom, R.S.; Delrow, J.; Bonson, P.; Burrack, A.L.; Hingorani, S.R.; Greenberg, P.D. Insufficiency of compound immune checkpoint blockade to overcome engineered T cell exhaustion in pancreatic cancer. J. Immunother. Cancer 2022, 10, e003525.
- Beatty, G.L.; Eghbali, S.; Kim, R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 267–278.
- Kemp, S.B.; Carpenter, E.S.; Steele, N.G.; Donahue, K.L.; Nwosu, Z.C.; Pacheco, A.; Velez-Delgado, A.; Menjivar, R.E.; Lima, F.; The, S.; et al. Apolipoprotein E Promotes Immune Suppression in Pancreatic Cancer through NF-kappaB-Mediated Production of CXCL1. Cancer Res. 2021, 81, 4305–4318.
- Long, K.B.; Collier, A.I.; Beatty, G.L. Macrophages: Key orchestrators of a tumor microenvironment defined by therapeutic resistance. Mol. Immunol. 2019, 110, 3–12.
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Li, T.; Liu, J.; Cui, M.; Zhang, X.; Gao, X.; Liao, Q.; et al. CXCL5 overexpression predicts a poor prognosis in pancreatic ductal adenocarcinoma and is correlated with immune cell infiltration. J. Cancer 2020, 11, 2371–2381.
- D’Errico, G.; Alonso-Nocelo, M.; Vallespinos, M.; Hermann, P.C.; Alcala, S.; Garcia, C.P.; Martin-Hijano, L.; Valle, S.; Earl, J.; Cassiano, C.; et al. Tumor-associated macrophage-secreted 14–3-3zeta signals via AXL to promote pancreatic cancer chemoresistance. Oncogene 2019, 38, 5469–5485.
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327.
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20.
- Di Caro, G.; Cortese, N.; Castino, G.F.; Grizzi, F.; Gavazzi, F.; Ridolfi, C.; Capretti, G.; Mineri, R.; Todoric, J.; Zerbi, A.; et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 2016, 65, 1710–1720.
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49.
- Hou, Y.C.; Chao, Y.J.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777.
- Singh, A.; Talekar, M.; Raikar, A.; Amiji, M. Macrophage-targeted delivery systems for nucleic acid therapy of inflammatory diseases. J. Control. Release 2014, 190, 515–530.
- Wang, J.; Cao, Z.; Zhang, X.M.; Nakamura, M.; Sun, M.; Hartman, J.; Harris, R.A.; Sun, Y.; Cao, Y. Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 2015, 75, 306–315.
- Habtezion, A.; Edderkaoui, M.; Pandol, S.J. Macrophages and pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 381, 211–216.
- Vayrynen, S.A.; Zhang, J.; Yuan, C.; Vayrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081.
- Bachy, S.; Wu, Z.; Gamradt, P.; Thierry, K.; Milani, P.; Chlasta, J.; Hennino, A. betaig-h3-structured collagen alters macrophage phenotype and function in pancreatic cancer. iScience 2022, 25, 103758.
- Farajzadeh Valilou, S.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61.
- Foucher, E.D.; Ghigo, C.; Chouaib, S.; Galon, J.; Iovanna, J.; Olive, D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front. Immunol. 2018, 9, 1044.
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286.
- Hu, H.; Hang, J.J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.W. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016, 37, 8657–8664.
- Yang, X.; Lin, J.; Wang, G.; Xu, D. Targeting Proliferating Tumor-Infiltrating Macrophages Facilitates Spatial Redistribution of CD8(+) T Cells in Pancreatic Cancer. Cancers 2022, 14, 1474.
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell. Immunol. 2017, 316, 1–10.
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Kruger, U.; Becker, T.; Ebsen, M.; Rocken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861.
- Li, C.; Cui, L.; Yang, L.; Wang, B.; Zhuo, Y.; Zhang, L.; Wang, X.; Zhang, Q.; Zhang, S. Pancreatic Stellate Cells Promote Tumor Progression by Promoting an Immunosuppressive Microenvironment in Murine Models of Pancreatic Cancer. Pancreas 2020, 49, 120–127.
- Bulle, A.; Dekervel, J.; Deschuttere, L.; Nittner, D.; Libbrecht, L.; Janky, R.; Plaisance, S.; Topal, B.; Coosemans, A.; Lambrechts, D.; et al. Gemcitabine Recruits M2-Type Tumor-Associated Macrophages into the Stroma of Pancreatic Cancer. Transl. Oncol. 2020, 13, 100743.
- Zhang, Y.; Yan, W.; Mathew, E.; Kane, K.T.; Brannon, A., 3rd; Adoumie, M.; Vinta, A.; Crawford, H.C.; Pasca di Magliano, M. Epithelial-Myeloid cell crosstalk regulates acinar cell plasticity and pancreatic remodeling in mice. Elife 2017, 6, e27388.
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083.
- Liu, Y.H.; Hu, C.M.; Hsu, Y.S.; Lee, W.H. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 817.
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640.
- Boyer, S.; Lee, H.J.; Steele, N.; Zhang, L.; Sajjakulnukit, P.; Andren, A.; Ward, M.H.; Singh, R.; Basrur, V.; Zhang, Y.; et al. Multiomic characterization of pancreatic cancer-associated macrophage polarization reveals deregulated metabolic programs driven by the GM-CSF-PI3K pathway. Elife 2022, 11, e73796.
- Legoffic, A.; Calvo, E.; Cano, C.; Folch-Puy, E.; Barthet, M.; Delpero, J.R.; Ferres-Maso, M.; Dagorn, J.C.; Closa, D.; Iovanna, J. The reg4 gene, amplified in the early stages of pancreatic cancer development, is a promising therapeutic target. PLoS ONE 2009, 4, e7495.
- He, X.J.; Jiang, X.T.; Ma, Y.Y.; Xia, Y.J.; Wang, H.J.; Guan, T.P.; Shao, Q.S.; Tao, H.Q. REG4 contributes to the invasiveness of pancreatic cancer by upregulating MMP-7 and MMP-9. Cancer Sci. 2012, 103, 2082–2091.
- Ma, X.; Wu, D.; Zhou, S.; Wan, F.; Liu, H.; Xu, X.; Xu, X.; Zhao, Y.; Tang, M. The pancreatic cancer secreted REG4 promotes macrophage polarization to M2 through EGFR/AKT/CREB pathway. Oncol. Rep. 2016, 35, 189–196.
- Sun, L.; Zhang, X.; Song, Q.; Liu, L.; Forbes, E.; Tian, W.; Zhang, Z.; Kang, Y.; Wang, H.; Fleming, J.B.; et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Lett. 2021, 500, 132–146.
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455.
- Chandrakesan, P.; Panneerselvam, J.; May, R.; Weygant, N.; Qu, D.; Berry, W.R.; Pitts, K.; Stanger, B.Z.; Rao, C.V.; Bronze, M.S.; et al. DCLK1-Isoform2 Alternative Splice Variant Promotes Pancreatic Tumor Immunosuppressive M2-Macrophage Polarization. Mol. Cancer Ther. 2020, 19, 1539–1549.
- Buyuk, B.; Jin, S.; Ye, K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell. Mol. Bioeng. 2022, 15, 1–13.
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Ochi, A.; Heindel, D.W.; Lee, K.B.; Zambirinis, C.P.; Pandian, G.S.B.; Savadkar, S.; et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat. Med. 2017, 23, 556–567.
- Seifert, A.M.; Reiche, C.; Heiduk, M.; Tannert, A.; Meinecke, A.C.; Baier, S.; von Renesse, J.; Kahlert, C.; Distler, M.; Welsch, T.; et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene 2020, 39, 3102–3113.
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546.
- Piao, J.; Liu, S.; Xu, Y.; Wang, C.; Lin, Z.; Qin, Y.; Liu, S. Ezrin protein overexpression predicts the poor prognosis of pancreatic ductal adenocarcinomas. Exp. Mol. Pathol. 2015, 98, 1–6.
- Chang, Y.T.; Peng, H.Y.; Hu, C.M.; Huang, S.C.; Tien, S.C.; Jeng, Y.M. Pancreatic cancer-derived small extracellular vesical Ezrin regulates macrophage polarization and promotes metastasis. Am. J. Cancer Res. 2020, 10, 12–37.
- Ghaffari, A.; Hoskin, V.; Szeto, A.; Hum, M.; Liaghati, N.; Nakatsu, K.; LeBrun, D.; Madarnas, Y.; Sengupta, S.; Elliott, B.E. A novel role for ezrin in breast cancer angio/lymphangiogenesis. Breast Cancer Res. 2014, 16, 438.
- Choi, J.W.; Kwon, M.J.; Kim, I.H.; Kim, Y.M.; Lee, M.K.; Nam, T.J. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int. J. Mol. Med. 2016, 38, 666–674.
- Su, M.J.; Aldawsari, H.; Amiji, M. Pancreatic Cancer Cell Exosome-Mediated Macrophage Reprogramming and the Role of MicroRNAs 155 and 125b2 Transfection using Nanoparticle Delivery Systems. Sci. Rep. 2016, 6, 30110.
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470.
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914.
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272.
- Yang, Y.; Andersson, P.; Hosaka, K.; Zhang, Y.; Cao, R.; Iwamoto, H.; Yang, X.; Nakamura, M.; Wang, J.; Zhuang, R.; et al. The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat. Commun. 2016, 7, 11385.
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3, e122375.
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2022, 71, 129–147.
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430.
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A., 3rd; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/mTOR Pathway in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 301–311.
- El Chartouni, C.; Schwarzfischer, L.; Rehli, M. Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology 2010, 215, 821–825.
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944.
- Bastea, L.I.; Liou, G.Y.; Pandey, V.; Fleming, A.K.; von Roemeling, C.A.; Doeppler, H.; Li, Z.; Qiu, Y.; Edenfield, B.; Copland, J.A.; et al. Pomalidomide Alters Pancreatic Macrophage Populations to Generate an Immune-Responsive Environment at Precancerous and Cancerous Lesions. Cancer Res. 2019, 79, 1535–1548.
- Lu, S.W.; Pan, H.C.; Hsu, Y.H.; Chang, K.C.; Wu, L.W.; Chen, W.Y.; Chang, M.S. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 2020, 11, 4611.
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738.
- Li, T.J.; Jin, K.Z.; Li, H.; Ye, L.Y.; Li, P.C.; Jiang, B.; Lin, X.; Liao, Z.Y.; Zhang, H.R.; Shi, S.M.; et al. SIGLEC15 amplifies immunosuppressive properties of tumor-associated macrophages in pancreatic cancer. Cancer Lett. 2022, 530, 142–155.
- Humphrey, M.B.; Lanier, L.L.; Nakamura, M.C. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol. Rev. 2005, 208, 50–65.
- Joshi, S.; Liu, K.X.; Zulcic, M.; Singh, A.R.; Skola, D.; Glass, C.K.; Sanders, P.D.; Sharabi, A.B.; Pham, T.V.; Tamayo, P.; et al. Macrophage Syk-PI3Kgamma Inhibits Antitumor Immunity: SRX3207, a Novel Dual Syk-PI3K Inhibitory Chemotype Relieves Tumor Immunosuppression. Mol. Cancer Ther. 2020, 19, 755–764.
- Lin, C.; He, H.; Liu, H.; Li, R.; Chen, Y.; Qi, Y.; Jiang, Q.; Chen, L.; Zhang, P.; Zhang, H.; et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 2019, 68, 1764–1773.
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41.
- Zhang, H.; Ye, Y.L.; Li, M.X.; Ye, S.B.; Huang, W.R.; Cai, T.T.; He, J.; Peng, J.Y.; Duan, T.H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2017, 36, 2095–2104.
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415.
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123.
- Diehl, L.; den Boer, A.T.; Schoenberger, S.P.; van der Voort, E.I.; Schumacher, T.N.; Melief, C.J.; Offringa, R.; Toes, R.E. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999, 5, 774–779.
- French, R.R.; Chan, H.T.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553.
- van Mierlo, G.J.; den Boer, A.T.; Medema, J.P.; van der Voort, E.I.; Fransen, M.F.; Offringa, R.; Melief, C.J.; Toes, R.E. CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc. Natl. Acad. Sci. USA 2002, 99, 5561–5566.
- Yasmin-Karim, S.; Bruck, P.T.; Moreau, M.; Kunjachan, S.; Chen, G.Z.; Kumar, R.; Grabow, S.; Dougan, S.K.; Ngwa, W. Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Front. Immunol. 2018, 9, 2030.
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58.
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586.
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411.
- Hezaveh, K.; Shinde, R.S.; Klotgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.
- Shinde, R.; McGaha, T.L. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends Immunol. 2018, 39, 1005–1020.
- Franchini, A.M.; Myers, J.R.; Jin, G.B.; Shepherd, D.M.; Lawrence, B.P. Genome-Wide Transcriptional Analysis Reveals Novel AhR Targets That Regulate Dendritic Cell Function during Influenza A Virus Infection. Immunohorizons 2019, 3, 219–235.
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728.
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935.
- Lofstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Ovenberger, M.; Poellinger, L.; Pahlman, S. Hypoxia inducible factor-2alpha in cancer. Cell Cycle 2007, 6, 919–926.
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634.
- Wahlgren, J.; De, L.K.T.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130.
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kgamma to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598.
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37.
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis-a key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369.
- Xiao, Z.; Li, J.; Yu, Q.; Zhou, T.; Duan, J.; Yang, Z.; Liu, C.; Xu, F. An Inflammatory Response Related Gene Signature Associated with Survival Outcome and Gemcitabine Response in Patients with Pancreatic Ductal Adenocarcinoma. Front. Pharmacol. 2021, 12, 778294.
- Ding, J.; He, X.; Cheng, X.; Cao, G.; Chen, B.; Chen, S.; Xiong, M. A 4-gene-based hypoxia signature is associated with tumor immune microenvironment and predicts the prognosis of pancreatic cancer patients. World J. Surg. Oncol. 2021, 19, 123.
- Song, W.; He, X.; Gong, P.; Yang, Y.; Huang, S.; Zeng, Y.; Wei, L.; Zhang, J. Glycolysis-Related Gene Expression Profiling Screen for Prognostic Risk Signature of Pancreatic Ductal Adenocarcinoma. Front. Genet. 2021, 12, 639246.
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031.
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898.
- Liu, Y.; Wang, X.; Zhu, Y.; Cao, Y.; Wang, L.; Li, F.; Zhang, Y.; Li, Y.; Zhang, Z.; Luo, J.; et al. The CTCF/LncRNA-PACERR complex recruits E1A binding protein p300 to induce pro-tumour macrophages in pancreatic ductal adenocarcinoma via directly regulating PTGS2 expression. Clin. Transl. Med. 2022, 12, e654.
- Edderkaoui, M.; Chheda, C.; Soufi, B.; Zayou, F.; Hu, R.W.; Ramanujan, V.K.; Pan, X.; Boros, L.G.; Tajbakhsh, J.; Madhav, A.; et al. An Inhibitor of GSK3B and HDACs Kills Pancreatic Cancer Cells and Slows Pancreatic Tumor Growth and Metastasis in Mice. Gastroenterology 2018, 155, 1985–1998.
This entry is offline, you can click here to edit this entry!