Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Primary familial brain calcification (PFBC), also known as Fahr’s disease, is a rare inherited disorder characterized by bilateral calcification in the basal ganglia according to neuroimaging. Other brain regions, such as the thalamus, cerebellum, and subcortical white matter, can also be affected. Among the diverse clinical phenotypes, the most common manifestations are movement disorders, cognitive deficits, and psychiatric disturbances. Although patients with PFBC always exhibit brain calcification, nearly one-third of cases remain clinically asymptomatic.
- primary familial brain calcification
- SLC20A2
- PDGFRB
- PDGFB
- XPR1
- MYORG
- JAM2
- CMPK2
1. Introduction
To date, two inheritance patterns have been found in PFBC patients. Heterozygous variants in SLC20A2, PDGFRB, PDGFB, and XPR1 are responsible for autosomal dominant PFBC [1][2][3][4], while biallelic changes in MYORG, JAM2, and CMPK2 are associated with autosomal recessive forms of the disease [5][6][7][8]. At present, around 50% of patients with PFBC do not have a pathogenic variant in the seven currently known genes [9], which indicates a more diverse genetic heterogeneity.
PFBC is genetically heterogeneous. To date, seven genes have been associated with PFBC, including four dominant genes and three recessive genes (Table 1).
Table 1. Summary of PFBC-causative genes.
| Gene | Locus | Mode of Inheritance | Protein | Expression | Function | Effect of Variant | Most Common Variant Type | References |
|---|---|---|---|---|---|---|---|---|
| SLC20A2 | Chr 8 | AD | Type III sodium-dependent inorganic phosphate transporter 2 (PiT2) | Ubiquitously, higher level in the brain | Uptake of inorganic phosphate (Pi) into cells | Loss of function | Missense | [1][10][11][12] |
| PDGFRB | Chr 5 | AD | Platelet-derived growth factor receptor-β (PDGFRB) | Neurons, vascular smooth muscle cells (SCMs), pericytes in the brain | Cell-surface tyrosine kinase receptors for the PDGF family, especially for homodimeric PDGF-B and PDGF-D; Essential for angiogenesis and formation of the blood–brain barrier (BBB) | Loss of function | Only missense | [2][9][10][13] |
| PDGFB | Chr 22 | AD | Platelet-derived growth factor subunit B (PDGFB) | Neurons and endothelial cells in the brain | Growth factors for mesenchymal cells; Crucial role in the proliferation and recruitment of pericytes and vascular SCMs | Loss of function | Missense | [3][10][14][15] |
| XPR1 | Chr 1 | AD | Xenotropic and polytropic retrovirus receptor 1 (XPR1) | Ubiquitously, higher level in the brain | Pi efflux from cells | Loss of function | Missense | [4][10][16] |
| MYORG | Chr 9 | AR | Myogenesis regulating glycosidase (MYORG) | Endoplasmic reticulum of the astrocytes in the brain | Member of the glycosyl hydrolase 31 family; Regulate protein glycosylation | Loss of function | Missense | [5][10] |
| JAM2 | Chr 21 | AR | Junctional-adhesion-molecule-2 (JAM2) | Endothelial cells and astrocytes in the brain | Member of the junctional adhesion molecules family; Crucial role in the regulation of cell polarity, endothelium permeability, leukocyte migration, and BBB function | Loss of function | Nonsense | [6][7][10] |
| CMPK2 | Chr 2 | AR | Uridine monophosphate-cytidine monophosphate kinase 2 (UMP-CMPK2) | Neurons and endothelial cells in the brain | Takes part in the salvage pathway for phosphorylation of dCMP, dUMP, CMP, and UMP in the mitochondria | Loss of function | Missense and start-codon loss | [8][17] |
Abbreviations: Chr: chromosome; AD: autosomal dominant; AR: autosomal recessive.
2. SLC20A2
The first PFBC-causative gene, SLC20A2, was identified in 2012 [1]. It is located on chromosome 8 and encodes for type III sodium-dependent inorganic phosphate (Pi) transporter 2 (PiT2). PiT2 has 12 transmembrane domains, and this protein is responsible for the uptake of Pi into cells [1][9][18]. SLC20A2 is expressed ubiquitously, but at a higher level in the brain, especially in the globus pallidus, thalamus, and cerebellum [11]. SLC20A2 is the most common PFBC gene; heterozygous variants have been identified in more than 60% of genetically confirmed PFBC patients [10]. A missense change is the most common variant type, followed by frameshift, nonsense, and splice site variations, without obvious hotspots for pathogenic variants (Figure 1a) [1][2][10][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61]. Functionally, both haploinsufficiency and dominant negative effects have been described; the loss of normal PiT2 function results in extracellular Pi accumulation and subsequent calcium phosphate formation [1][12]. Calcification around the blood vessels or within the vessel walls of involved brain regions has been demonstrated in Slc20a2 homozygous knockout mice (Slc20a2−/− mice) and in an autopsied SLC20A2-PFBC patient [62][63][64]. Calcification in Slc20a2−/− mice was also found inside cells, mainly in the pericytes and astrocytes, which suggested the intracellular cytosolic initiation of calcification [63]. Moreover, increasing T-cell infiltration in the brain parenchyma was found in Slc20a2−/− mice, which is positively associated with brain calcification and aging. Impaired blood–brain barrier (BBB) permeability with the enhancement of endocytosis and transcytosis was also demonstrated, which may be explained by the dysfunction of pericytes and astrocytes due to intracellular calcification [65]. In addition, PiT2 is known to be expressed in the apical membrane of choroid plexus epithelial cells in spiny dogfish and rats, suggesting that PiT2 plays an important role in actively transporting Pi from the cerebrospinal fluid (CSF) to the blood to maintain phosphate homeostasis in the CSF [66]. The level of Pi in CSF is significantly elevated in both Slc20a2 homozygous knockout mice and PFBC patients with SLC20A2 pathogenic variants [63][67][68]. In summary, PiT2 dysfunction can leads to a local increase in the extracellular and CSF Pi concentrations, which then trigger cell-mediated mineralization progression, ensuing calcification [62][63].
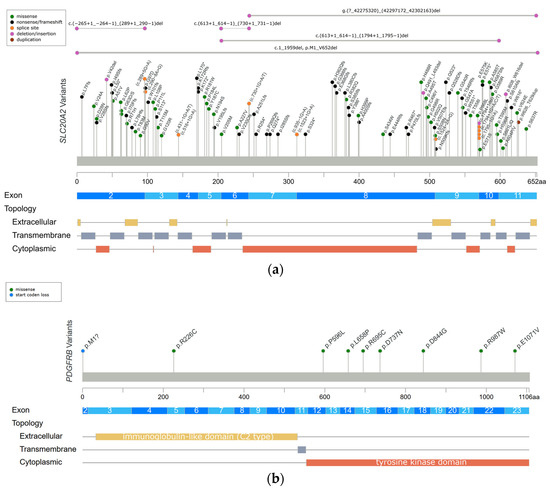
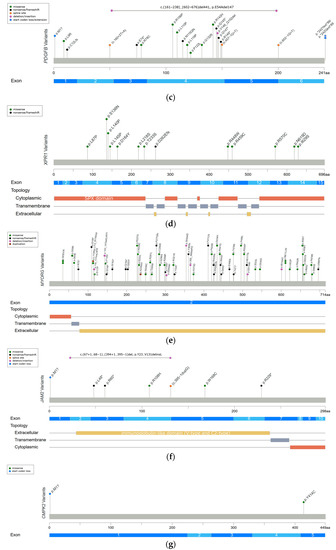
Figure 1. Reported variants in seven genes linked to PFBC along the protein sequence and their topologic protein models. (a) The reported variants in the SLC20A2 gene and topology protein model of PiT2. (b) PDGFRB gene and PDGF-Rβ. (c) PDGFB gene. (d) XPR1 gene and XPR1. (e) MYORG gene and MYORG. (f) JAM2 gene and JAM2. (g) CMPK2 gene. There is no obvious hotspot for SLC20A2, MYORG, and JAM2 genes. The variants tend to cluster in the tyrosine kinase domain of the PDGFRB gene, the mature protein product between positions 82 and 190 of the PDGFB gene, and in the SPX domain of the XPR1 gene. Meaning of symbols: *, stop codon; ?, unknown (a variant affecting the initiation codon cannot be predicted).
3. PDGFRB
The PDGFRB gene was identified in PFBC patients in 2013 [2], and it is located on chromosome 5 and encodes for platelet-derived growth factor receptor-β (PDGF-Rβ). The structure of PDGF-Rβ includes five extracellular immunoglobulin-like C2 type domains, a transmembrane domain, and a tyrosine kinase domain. PDGF-Rβ is a cell-surface tyrosine kinase receptor for members of the platelet-derived growth factor (PDGF) family, with a high affinity for homodimeric PDGF-B and PDGF-D. At the tissue level, the PDGFRB gene is highly expressed in the brain, especially in the basal ganglia and dentate nucleus of the cerebellum. At the cellular level, the PDGFRB gene is expressed in neurons, vascular smooth muscle cells (SCMs), and pericytes. The signal transduction of PDGF-Rβ and its ligand is essential for the proliferation and migration of vascular SCMs and pericytes, and subsequently, the angiogenesis and formation of the BBB [2][9][18]. Heterozygous variants in the PDGFRB gene have been identified in 5% of genetically confirmed PFBC patients [10]. The missense change is currently the only variant type to be identified in the PDGFRB gene. The variants tend to cluster in the tyrosine kinase domain (Figure 1b) [2][10][21][32][69][70], and they are likely to cause haploinsufficiency and affect the kinase function of the protein [13]. The functional loss of PDGF-Rβ may lead to pericytes dysfunction, which would impact BBB integrity and secondarily induce calcium depositions in the vessel walls or perivascular space. Several studies have shown that homodimeric PDGF-B can directly induce vascular SCM calcification via enhancing the expression of inorganic phosphate transporter 1 (PiT1) [71][72]. Hence, Nicolas et al. hypothesized that activating variants in PDGFRB may impact the PDGF-PiT1 pathway and cause vascular calcification [2]. Interestingly, PiT1 is encoded by the SLC20A1 gene, which is one of two type III sodium-dependent Pi transporters. The other is PiT2 encoded by SLC20A2 [2][70]. However, there is no further evidence supporting this hypothesis [73].
4. PDGFB
The PDGFB gene was also reported in 2013 [3], and it is located on chromosome 22 and encodes for the PDGF-B precursor protein. This precursor protein is cleaved at positions 81 and 191, and subsequently forms a homodimer through disulfide bonds, which is the main ligand of PDGF-Rβ [18]. The PDGFB gene is expressed in neurons and endothelial cells in the brain [3]. PDGF-B is a growth factor for mesenchymal cells and plays a crucial role in the proliferation and recruitment of pericytes and vascular SMCs. [14][15]. Heterozygous variants in the PDGFB gene have been identified in 12% of genetically confirmed PFBC patients [10]. The missense change is the most common variant type in the PDGFB gene, followed by nonsense, splice site, and extension variants. The variants cluster between protein positions 82 and 190, which are retained in the mature PDGF-B protein (Figure 1c) [3][10][19][32][35][45][58][74][75][76][77][78][79][80][81][82]. The same as PDGFRB does, PDGFB variants also cause haploinsufficiency, either by deleting critical parts of protein or disrupting normal protein function. The loss of normal PDGF-B function leads to BBB impairment via PDGF-Rβ, and then triggers the process of calcification [3][9].
5. XPR1
The XPR1 gene was identified in 2015 [4], and it is located on chromosome 1 and encodes for xenotropic and polytropic retrovirus receptor 1 (XPR1). XPR contains eight transmembrane domains and an amino-terminal SPX domain [18]. This protein mediates Pi efflux from cells [4][9]. XPR1 is expressed universally, and a high XPR1 mRNA level has been demonstrated in mouse brains, especially in the cerebellum and striatum [16]. Heterozygous variants in the XPR1 gene have been identified in 6% of genetically confirmed PFBC patients [10]. The missense change is the most common variant in XPR1-related PFBC patients, which tends to cluster in the SPX domain (Figure 1d) [4][10][29][32][83][84][85]. XPR1 variants may cause haploinsufficiency, leading to the intracellular accumulation of Pi and formation of calcium phosphate [4]. Interestingly, mutual interactions between XPR1 and PDGFRB were found in a recent immunoprecipitation study [16]. It is hypothesized that these two proteins may form a complex on the cell membrane, further suggesting that PDGFRB may be the upstream regulator of XPR1 [16].
6. MYORG
In 2018, the first and most common autosomal recessive PFBC-causative gene, MYORG, was identified. MYORG is located on chromosome 9 and encodes for myogenesis-regulating glycosidase (MYORG). It contains a short cytoplasm domain at the N-terminal, a transmembrane domain, and a long luminal fragment with a glycosidase domain at the C-terminal. MYORG is a member of the glycosyl hydrolase 31 family, and its function is to regulate protein glycosylation. In the brain, the MYORG gene is highly expressed in the cerebellum, specifically in the endoplasmic reticulum of the astrocytes [5]. Biallelic variants in the MYORG gene have been identified in 13% of genetically confirmed PFBC patients [10]. The missense change is the most common variant type in the MYORG gene, followed by in-frame indels, nonsense, and frameshift variations. There is no obvious hotspot of pathogenic variants (Figure 1e) [5][10][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102]. Pathogenic variants cause the loss of the glycosidase function of MYORG, which may lead to abnormal protein glycosylation and metabolic disturbance. It is believed that MYORG variants can induce astrocyte dysfunction, which then disturbs the association between astrocytes and pericytes, resulting in neurovascular unit (NVU) dysfunction and subsequently causing the formation of brain calcification [5]. However, the exact linkage between the loss of protein glycosylation and astrocyte dysfunction remains to be elucidated.
7. JAM2
In 2020, another autosomal recessive PFBC-causative gene, JAM2, was identified. JAM2 is located on chromosome 21 and encodes for junctional-adhesion-molecule-2 (JAM2). The structure of JAM2 includes two immunoglobulin-like domains (V-type and C2-type). JAM2 is a member of the junctional adhesion molecule family, and it plays crucial roles in the regulation of cell polarity, endothelium permeability, leukocyte migration, and BBB function [6][7]. In the brain, JAM2 is highly expressed in the caudate nuclei. At the cellular level, JAM2 is specifically expressed in endothelial cells and astrocytes [7]. Biallelic variants in the JAM2 gene have been identified in 2% of genetically confirmed PFBC patients [10]. The nonsense change is the most common variant type in the JAM2 gene. Missense, frameshift, and structural variants have also been reported, without a mutation hotspot (Figure 1f) [6][7][10]. Variants of JAM2 gene cause the loss of cell–cell adhesion and the dysfunction of the solute passage, which may contribute to the formation of brain calcification [6][7].
8. CMPK2
The CMPK2 gene is the latest autosomal recessive PFBC-causative gene to be identified at the end of 2022 [8]. This gene is located on chromosome 2 and encodes for uridine monophosphate-cytidine monophosphate kinase 2 (UMP-CMPK2), which can be separated into N-terminal and C-terminal domains according to the sequence properties. CMPK2 is a member of the nucleoside monophosphate kinase family and participates in the salvage pathway for the phosphorylation of dCMP, dUMP, CMP, and UMP in the mitochondria [17]. In the brain, the CMPK2 gene is highly expressed in the hippocampus and cerebellum. At the cellular level, CMPK2 is specifically expressed in neurons and vascular endothelial cells [8]. To date, biallelic variants in the CMPK2 gene have only been reported in two PFBC families, which carry missense and start-codon loss variants (Figure 1g) [8]. The loss of UMP-CMPK2 function leads to a reduction in mitochondrial genome DNA copy numbers, the downregulation of the expression of mitochondrial protein, the decrease in ATP production, and the disturbance of mitochondrial cristae morphology. The disturbance of mitochondrial function is believed to cause impairment of energy homeostasis and the upregulation of intracellular phosphate levels, subsequently triggering the development of brain calcification [8].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241310886
References
- Wang, C.; Li, Y.; Shi, L.; Ren, J.; Patti, M.; Wang, T.; de Oliveira, J.R.; Sobrido, M.J.; Quintans, B.; Baquero, M.; et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 2012, 44, 254–256.
- Nicolas, G.; Pottier, C.; Maltete, D.; Coutant, S.; Rovelet-Lecrux, A.; Legallic, S.; Rousseau, S.; Vaschalde, Y.; Guyant-Marechal, L.; Augustin, J.; et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 2013, 80, 181–187.
- Keller, A.; Westenberger, A.; Sobrido, M.J.; Garcia-Murias, M.; Domingo, A.; Sears, R.L.; Lemos, R.R.; Ordonez-Ugalde, A.; Nicolas, G.; da Cunha, J.E.; et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 2013, 45, 1077–1082.
- Legati, A.; Giovannini, D.; Nicolas, G.; Lopez-Sanchez, U.; Quintans, B.; Oliveira, J.R.; Sears, R.L.; Ramos, E.M.; Spiteri, E.; Sobrido, M.J.; et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 2015, 47, 579–581.
- Yao, X.P.; Cheng, X.; Wang, C.; Zhao, M.; Guo, X.X.; Su, H.Z.; Lai, L.L.; Zou, X.H.; Chen, X.J.; Zhao, Y.; et al. Biallelic Mutations in MYORG Cause Autosomal Recessive Primary Familial Brain Calcification. Neuron 2018, 98, 1116–1123 e1115.
- Schottlaender, L.V.; Abeti, R.; Jaunmuktane, Z.; Macmillan, C.; Chelban, V.; O’Callaghan, B.; McKinley, J.; Maroofian, R.; Efthymiou, S.; Athanasiou-Fragkouli, A.; et al. Bi-allelic JAM2 Variants Lead to Early-Onset Recessive Primary Familial Brain Calcification. Am. J. Hum. Genet. 2020, 106, 412–421.
- Cen, Z.; Chen, Y.; Chen, S.; Wang, H.; Yang, D.; Zhang, H.; Wu, H.; Wang, L.; Tang, S.; Ye, J.; et al. Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain 2020, 143, 491–502.
- Zhao, M.; Su, H.Z.; Zeng, Y.H.; Sun, Y.; Guo, X.X.; Li, Y.L.; Wang, C.; Zhao, Z.Y.; Huang, X.J.; Lin, K.J.; et al. Loss of function of CMPK2 causes mitochondria deficiency and brain calcification. Cell Discov. 2022, 8, 128.
- Westenberger, A.; Balck, A.; Klein, C. Primary familial brain calcifications: Genetic and clinical update. Curr. Opin. Neurol. 2019, 32, 571–578.
- Balck, A.; Schaake, S.; Kuhnke, N.S.; Domingo, A.; Madoev, H.; Margolesky, J.; Dobricic, V.; Alvarez-Fischer, D.; Laabs, B.H.; Kasten, M.; et al. Genotype-Phenotype Relations in Primary Familial Brain Calcification: Systematic MDSGene Review. Mov. Disord. 2021, 36, 2468–2480.
- da Silva, R.J.; Pereira, I.C.; Oliveira, J.R. Analysis of gene expression pattern and neuroanatomical correlates for SLC20A2 (PiT-2) shows a molecular network with potential impact in idiopathic basal ganglia calcification (“Fahr’s disease”). J. Mol. Neurosci. 2013, 50, 280–283.
- Larsen, F.T.; Jensen, N.; Autzen, J.K.; Kongsfelt, I.B.; Pedersen, L. Primary Brain Calcification Causal PiT2 Transport-Knockout Variants can Exert Dominant Negative Effects on Wild-Type PiT2 Transport Function in Mammalian Cells. J. Mol. Neurosci. 2017, 61, 215–220.
- Arts, F.A.; Velghe, A.I.; Stevens, M.; Renauld, J.C.; Essaghir, A.; Demoulin, J.B. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J. Cell. Mol. Med. 2015, 19, 239–248.
- Vanlandewijck, M.; Lebouvier, T.; Andaloussi Mae, M.; Nahar, K.; Hornemann, S.; Kenkel, D.; Cunha, S.I.; Lennartsson, J.; Boss, A.; Heldin, C.H.; et al. Functional Characterization of Germline Mutations in PDGFB and PDGFRB in Primary Familial Brain Calcification. PLoS ONE 2015, 10, e0143407.
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312.
- Yao, X.P.; Zhao, M.; Wang, C.; Guo, X.X.; Su, H.Z.; Dong, E.L.; Chen, H.T.; Lai, J.H.; Liu, Y.B.; Wang, N.; et al. Analysis of gene expression and functional characterization of XPR1: A pathogenic gene for primary familial brain calcification. Cell Tissue Res. 2017, 370, 267–273.
- Xu, Y.; Johansson, M.; Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 2008, 283, 1563–1571.
- Quintans, B.; Oliveira, J.; Sobrido, M.J. Primary familial brain calcifications. Handb. Clin. Neurol. 2018, 147, 307–317.
- Zhan, F.X.; Tian, W.T.; Zhang, C.; Zhu, Z.Y.; Wang, S.G.; Huang, X.J.; Cao, L. Primary familial brain calcification presenting as paroxysmal kinesigenic dyskinesia: Genetic and functional analyses. Neurosci. Lett. 2020, 714, 134543.
- Zhu, M.; Zhu, X.; Wan, H.; Hong, D. Familial IBGC caused by SLC20A2 mutation presenting as paroxysmal kinesigenic dyskinesia. Park. Relat. Disord. 2014, 20, 353–354.
- Nicolas, G.; Pottier, C.; Charbonnier, C.; Guyant-Marechal, L.; Le Ber, I.; Pariente, J.; Labauge, P.; Ayrignac, X.; Defebvre, L.; Maltete, D.; et al. Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 2013, 136, 3395–3407.
- Zhang, X.; Ma, G.; Zhao, Z.; Zhu, M. SCL20A2 mutation presenting with acute ischemic stroke: A case report. BMC Neurol. 2018, 18, 11.
- Ferreira, J.B.; Pimentel, L.; Keasey, M.P.; Lemos, R.R.; Santos, L.M.; Oliveira, M.F.; Santos, S.; Jensen, N.; Teixeira, K.; Pedersen, L.; et al. First report of a de novo mutation at SLC20A2 in a patient with brain calcification. J. Mol. Neurosci. 2014, 54, 748–751.
- Baker, M.; Strongosky, A.J.; Sanchez-Contreras, M.Y.; Yang, S.; Ferguson, W.; Calne, D.B.; Calne, S.; Stoessl, A.J.; Allanson, J.E.; Broderick, D.F.; et al. SLC20A2 and THAP1 deletion in familial basal ganglia calcification with dystonia. Neurogenetics 2014, 15, 23–30.
- Batla, A.; Stamelou, M. Primary familial brain calcification in the IBGC2 kindred: All linkage roads lead to SLC20A2. Mov. Disord. 2016, 31, 1765–1766.
- Giorgio, E.; Garelli, E.; Carando, A.; Bellora, S.; Rubino, E.; Quarello, P.; Sirchia, F.; Marrama, F.; Gallone, S.; Grosso, E.; et al. Design of a multiplex ligation-dependent probe amplification assay for SLC20A2: Identification of two novel deletions in primary familial brain calcification. J. Hum. Genet. 2019, 64, 1083–1090.
- Guo, X.X.; Su, H.Z.; Zou, X.H.; Lai, L.L.; Lu, Y.Q.; Wang, C.; Li, Y.L.; Hong, J.M.; Zhao, M.; Lin, K.X.; et al. Identification of SLC20A2 deletions in patients with primary familial brain calcification. Clin. Genet. 2019, 96, 53–60.
- Gagliardi, M.; Morelli, M.; Annesi, G.; Nicoletti, G.; Perrotta, P.; Pustorino, G.; Iannello, G.; Tarantino, P.; Gambardella, A.; Quattrone, A. A new SLC20A2 mutation identified in southern Italy family with primary familial brain calcification. Gene 2015, 568, 109–111.
- Guo, X.X.; Zou, X.H.; Wang, C.; Yao, X.P.; Su, H.Z.; Lai, L.L.; Chen, H.T.; Lai, J.H.; Liu, Y.B.; Chen, D.P.; et al. Spectrum of SLC20A2, PDGFRB, PDGFB, and XPR1 mutations in a large cohort of patients with primary familial brain calcification. Hum. Mutat. 2019, 40, 392–403.
- Chen, W.J.; Yao, X.P.; Zhang, Q.J.; Ni, W.; He, J.; Li, H.F.; Liu, X.Y.; Zhao, G.X.; Murong, S.X.; Wang, N.; et al. Novel SLC20A2 mutations identified in southern Chinese patients with idiopathic basal ganglia calcification. Gene 2013, 529, 159–162.
- Nishii, K.; Shimogawa, R.; Kurita, H.; Inden, M.; Kobayashi, M.; Toyoshima, I.; Taguchi, Y.; Ueda, A.; Tamune, H.; Hozumi, I. Partial reduced Pi transport function of PiT-2 might not be sufficient to induce brain calcification of idiopathic basal ganglia calcification. Sci. Rep. 2019, 9, 17288.
- Ramos, E.M.; Carecchio, M.; Lemos, R.; Ferreira, J.; Legati, A.; Sears, R.L.; Hsu, S.C.; Panteghini, C.; Magistrelli, L.; Salsano, E.; et al. Primary brain calcification: An international study reporting novel variants and associated phenotypes. Eur. J. Hum. Genet. 2018, 26, 1462–1477.
- Yamada, M.; Tanaka, M.; Takagi, M.; Kobayashi, S.; Taguchi, Y.; Takashima, S.; Tanaka, K.; Touge, T.; Hatsuta, H.; Murayama, S.; et al. Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology 2014, 82, 705–712.
- Ding, Y.; Dong, H.Q. A Novel SLC20A2 Mutation Associated with Familial Idiopathic Basal Ganglia Calcification and Analysis of the Genotype-Phenotype Association in Chinese Patients. Chin. Med. J. 2018, 131, 799–803.
- Koyama, S.; Sato, H.; Kobayashi, R.; Kawakatsu, S.; Kurimura, M.; Wada, M.; Kawanami, T.; Kato, T. Clinical and radiological diversity in genetically confirmed primary familial brain calcification. Sci. Rep. 2017, 7, 12046.
- Magistrelli, L.; Croce, R.; De Marchi, F.; Basagni, C.; Carecchio, M.; Nasuelli, N.; Cantello, R.; Invernizzi, F.; Garavaglia, B.; Comi, C.; et al. Expanding the genetic spectrum of primary familial brain calcification due to SLC2OA2 mutations: A case series. Neurogenetics 2021, 22, 65–70.
- Kasuga, K.; Konno, T.; Saito, K.; Ishihara, A.; Nishizawa, M.; Ikeuchi, T. A Japanese family with idiopathic basal ganglia calcification with novel SLC20A2 mutation presenting with late-onset hallucination and delusion. J. Neurol. 2014, 261, 242–244.
- Carecchio, M.; Barzaghi, C.; Varrasi, C.; Cantello, R.; Garavaglia, B. Adult-Onset Focal Chorea in Fahr’s Disease Resulting From SLC20A2 Mutation: A Novel Phenotype. Mov. Disord. Clin. Pract. 2015, 2, 79–80.
- Hsu, S.C.; Sears, R.L.; Lemos, R.R.; Quintans, B.; Huang, A.; Spiteri, E.; Nevarez, L.; Mamah, C.; Zatz, M.; Pierce, K.D.; et al. Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 2013, 14, 11–22.
- Konno, T.; Blackburn, P.R.; Rozen, T.D.; van Gerpen, J.A.; Ross, O.A.; Atwal, P.S.; Wszolek, Z.K. Anticipation in a family with primary familial brain calcification caused by an SLC20A2 variant. Neurol. Neurochir. Pol. 2018, 52, 386–389.
- Zhang, Y.; Guo, X.; Wu, A. Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PLoS ONE 2013, 8, e57060.
- Lemos, R.R.; Ramos, E.M.; Legati, A.; Nicolas, G.; Jenkinson, E.M.; Livingston, J.H.; Crow, Y.J.; Campion, D.; Coppola, G.; Oliveira, J.R. Update and Mutational Analysis of SLC20A2: A Major Cause of Primary Familial Brain Calcification. Hum. Mutat. 2015, 36, 489–495.
- Zhang, Q.; Li, Z.; Sun, H.; Zhang, S.; Zhang, J.; Wang, Y.; Fang, H.; Xu, Y. Generation of induced pluripotent stem cell line (ZZUi0012-A) from a patient with Fahr’s disease caused by a novel mutation in SLC20A2 gene. Stem Cell Res. 2019, 35, 101395.
- Huang, Y.T.; Zhang, L.H.; Li, M.F.; Cheng, L.; Zou, G.Y.; Zhou, H.H. A splice site mutation causing exon 6 skipping in SLC20A2 gene in a primary familial brain calcification family. Brain Res. Bull. 2019, 150, 261–265.
- Shen, Y.; Shu, S.; Ren, Y.; Xia, W.; Chen, J.; Dong, L.; Ge, H.; Fan, S.; Shi, L.; Peng, B.; et al. Case Report: Two Novel Frameshift Mutations in SLC20A2 and One Novel Splice Donor Mutation in PDGFB Associated With Primary Familial Brain Calcification. Front. Genet. 2021, 12, 643452.
- Rohani, M.; Poon, Y.Y.; Naranian, T.; Fasano, A. SCL20A2 mutation mimicking fluctuating Parkinson’s disease. Park. Relat. Disord. 2017, 39, 93–94.
- Knowles, J.K.; Santoro, J.D.; Porter, B.E.; Baumer, F.M. Refractory focal epilepsy in a paediatric patient with primary familial brain calcification. Seizure 2018, 56, 50–52.
- Li, M.; Fu, Q.; Xiang, L.; Zheng, Y.; Ping, W.; Cao, Y. SLC20A2-Associated Idiopathic basal ganglia calcification (Fahr disease): A case family report. BMC Neurol. 2022, 22, 438.
- Taglia, I.; Mignarri, A.; Olgiati, S.; Menci, E.; Petrocelli, P.L.; Breedveld, G.J.; Scaglione, C.; Martinelli, P.; Federico, A.; Bonifati, V.; et al. Primary familial brain calcification: Genetic analysis and clinical spectrum. Mov. Disord. 2014, 29, 1691–1695.
- Paucar, M.; Almqvist, H.; Jelic, V.; Hagman, G.; Jorneskog, G.; Holmin, S.; Bjorkhem, I.; Svenningsson, P. A SLC20A2 gene mutation carrier displaying ataxia and increased levels of cerebrospinal fluid phosphate. J. Neurol. Sci. 2017, 375, 245–247.
- Lemos, R.R.; Oliveira, M.F.; Oliveira, J.R.M. Reporting a new mutation at the SLC20A2 gene in familial idiopathic basal ganglia calcification. Eur. J. Neurol. 2013, 20, e43–e44.
- Uno, A.; Tamune, H.; Kurita, H.; Hozumi, I.; Yamamoto, N. SLC20A2-Associated Idiopathic Basal Ganglia Calcification-Related Recurrent Psychosis Response to Low-Dose Antipsychotics: A Case Report and Literature Review. Cureus 2020, 12, e12407.
- Rubino, E.; Giorgio, E.; Gallone, S.; Pinessi, L.; Orsi, L.; Gentile, S.; Duca, S.; Brusco, A. Novel mutation of SLC20A2 in an Italian patient presenting with migraine. J. Neurol. 2014, 261, 2019–2021.
- Coppola, A.; Hernandez-Hernandez, L.; Balestrini, S.; Krithika, S.; Moran, N.; Hale, B.; Cordivari, C.; Sisodiya, S.M. Cortical myoclonus and epilepsy in a family with a new SLC20A2 mutation. J. Neurol. 2020, 267, 2221–2227.
- Brighina, L.; Saracchi, E.; Ferri, F.; Gagliardi, M.; Tarantino, P.; Morzenti, S.; Musarra, M.; Patassini, M.; Annesi, G.; Ferrarese, C. Fahr’s disease linked to a novel SLC20A2 gene mutation manifesting with dynamic aphasia. Neurodegener. Dis. 2014, 14, 133–138.
- Fjaer, R.; Brodtkorb, E.; Oye, A.M.; Sheng, Y.; Vigeland, M.D.; Kvistad, K.A.; Backe, P.H.; Selmer, K.K. Generalized epilepsy in a family with basal ganglia calcifications and mutations in SLC20A2 and CHRNB2. Eur. J. Med. Genet. 2015, 58, 624–628.
- Bu, W.; Hou, L.; Zhu, M.; Zhang, R.; Zhang, X.; Zhang, X.; Tang, J.; Liu, X. SLC20A2-related primary familial brain calcification with purely acute psychiatric symptoms: A case report. BMC Neurol. 2022, 22, 265.
- Sun, H.; Cao, Z.; Gao, R.; Li, Y.; Chen, R.; Du, S.; Ma, T.; Wang, J.; Xu, X.; Liu, J.Y. Severe brain calcification and migraine headache caused by SLC20A2 and PDGFRB heterozygous mutations in a five-year-old Chinese girl. Mol. Genet. Genom. Med. 2021, 9, e1670.
- Sekine, S.I.; Kondo, T.; Murakami, N.; Imamura, K.; Enami, T.; Shibukawa, R.; Tsukita, K.; Funayama, M.; Inden, M.; Kurita, H.; et al. Induced pluripotent stem cells derived from a patient with familial idiopathic basal ganglia calcification (IBGC) caused by a mutation in SLC20A2 gene. Stem Cell Res. 2017, 24, 40–43.
- Taglia, I.; Formichi, P.; Battisti, C.; Peppoloni, G.; Barghigiani, M.; Tessa, A.; Federico, A. Primary familial brain calcification with a novel SLC20A2 mutation: Analysis of PiT-2 expression and localization. J. Cell. Physiol. 2018, 233, 2324–2331.
- McKenna, M.C.; Redmond, J.; Bradley, D.; Bede, P. Teaching NeuroImage: Primary Familial Brain Calcification in SLC20A2 Genotype. Neurology 2022, 99, 1008–1009.
- Jensen, N.; Schroder, H.D.; Hejbol, E.K.; Fuchtbauer, E.M.; de Oliveira, J.R.; Pedersen, L. Loss of function of Slc20a2 associated with familial idiopathic Basal Ganglia calcification in humans causes brain calcifications in mice. J. Mol. Neurosci. 2013, 51, 994–999.
- Jensen, N.; Schroder, H.D.; Hejbol, E.K.; Thomsen, J.S.; Bruel, A.; Larsen, F.T.; Vinding, M.C.; Orlowski, D.; Fuchtbauer, E.M.; Oliveira, J.R.M.; et al. Mice Knocked Out for the Primary Brain Calcification-Associated Gene Slc20a2 Show Unimpaired Prenatal Survival but Retarded Growth and Nodules in the Brain that Grow and Calcify Over Time. Am. J. Pathol. 2018, 188, 1865–1881.
- Kimura, T.; Miura, T.; Aoki, K.; Saito, S.; Hondo, H.; Konno, T.; Uchiyama, A.; Ikeuchi, T.; Takahashi, H.; Kakita, A. Familial idiopathic basal ganglia calcification: Histopathologic features of an autopsied patient with an SLC20A2 mutation. Neuropathology 2016, 36, 365–371.
- Zhang, Y.; Ren, Y.; Zhang, Y.; Li, Y.; Xu, C.; Peng, Z.; Jia, Y.; Qiao, S.; Zhang, Z.; Shi, L. T-cell infiltration in the central nervous system and their association with brain calcification in Slc20a2-deficient mice. Front. Mol. Neurosci. 2023, 16, 1073723.
- Guerreiro, P.M.; Bataille, A.M.; Parker, S.L.; Renfro, J.L. Active removal of inorganic phosphate from cerebrospinal fluid by the choroid plexus. Am. J. Physiol. Renal. Physiol. 2014, 306, F1275–F1284.
- Jensen, N.; Autzen, J.K.; Pedersen, L. SLC20A2 is critical for maintaining a physiologic inorganic phosphate level in cerebrospinal fluid. Neurogenetics 2016, 17, 125–130.
- Hozumi, I.; Kurita, H.; Ozawa, K.; Furuta, N.; Inden, M.; Sekine, S.I.; Yamada, M.; Hayashi, Y.; Kimura, A.; Inuzuka, T.; et al. Inorganic phosphorus (Pi) in CSF is a biomarker for Slc20a2-associated idiopathic basal ganglia calcification (IBGC1). J. Neurol. Sci. 2018, 388, 150–154.
- Wang, C.; Yao, X.P.; Chen, H.T.; Lai, J.H.; Guo, X.X.; Su, H.Z.; Dong, E.L.; Zhang, Q.J.; Wang, N.; Chen, W.J. Novel mutations of PDGFRB cause primary familial brain calcification in Chinese families. J. Hum. Genet. 2017, 62, 697–701.
- Sanchez-Contreras, M.; Baker, M.C.; Finch, N.A.; Nicholson, A.; Wojtas, A.; Wszolek, Z.K.; Ross, O.A.; Dickson, D.W.; Rademakers, R. Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum. Mutat. 2014, 35, 964–971.
- Villa-Bellosta, R.; Levi, M.; Sorribas, V. Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: Effects of PDGF, TNF-alpha, and Pi. Pflug. Arch. 2009, 458, 1151–1161.
- Kakita, A.; Suzuki, A.; Nishiwaki, K.; Ono, Y.; Kotake, M.; Ariyoshi, Y.; Miura, Y.; Ltoh, M.; Oiso, Y. Stimulation of Na-dependent phosphate transport by platelet-derived growth factor in rat aortic smooth muscle cells. Atherosclerosis 2004, 174, 17–24.
- Xu, X.; Sun, H.; Luo, J.; Cheng, X.; Lv, W.; Luo, W.; Chen, W.J.; Xiong, Z.Q.; Liu, J.Y. The Pathology of Primary Familial Brain Calcification: Implications for Treatment. Neurosci. Bull. 2022, 39, 659–674.
- Sekine, S.I.; Kaneko, M.; Tanaka, M.; Ninomiya, Y.; Kurita, H.; Inden, M.; Yamada, M.; Hayashi, Y.; Inuzuka, T.; Mitsui, J.; et al. Functional evaluation of PDGFB-variants in idiopathic basal ganglia calcification, using patient-derived iPS cells. Sci. Rep. 2019, 9, 5698.
- Hayashi, T.; Legati, A.; Nishikawa, T.; Coppola, G. First Japanese family with primary familial brain calcification due to a mutation in the PDGFB gene: An exome analysis study. Psychiatry Clin. Neurosci. 2015, 69, 77–83.
- Nicolas, G.; Jacquin, A.; Thauvin-Robinet, C.; Rovelet-Lecrux, A.; Rouaud, O.; Pottier, C.; Aubriot-Lorton, M.H.; Rousseau, S.; Wallon, D.; Duvillard, C.; et al. A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur. J. Hum. Genet. 2014, 22, 1236–1238.
- Keogh, M.J.; Pyle, A.; Daud, D.; Griffin, H.; Douroudis, K.; Eglon, G.; Miller, J.; Horvath, R.; Chinnery, P.F. Clinical heterogeneity of primary familial brain calcification due to a novel mutation in PDGFB. Neurology 2015, 84, 1818–1820.
- Biyajima, M.; Kobayashi, Y.; Nakafuji, K.; Watanabe, R.; Tazawa, K.; Ishii, W.; Satoh, S.; Hoshi, K.; Kurita, H.; Hozumi, I.; et al. Seronegative neuromyelitis optica spectrum disorder in primary familial brain calcification with PDGFB variant. eNeurologicalSci 2022, 27, 100406.
- Yao, X.P.; Wang, C.; Su, H.Z.; Guo, X.X.; Lu, Y.Q.; Zhao, M.; Liu, Y.B.; Lai, J.H.; Chen, H.T.; Wang, N.; et al. Mutation screening of PDGFB gene in Chinese population with primary familial brain calcification. Gene 2016, 597, 17–22.
- Wang, C.; Ma, X.; Xu, X.; Huang, B.; Sun, H.; Li, L.; Zhang, M.; Liu, J.Y. A PDGFB mutation causes paroxysmal nonkinesigenic dyskinesia with brain calcification. Mov. Disord. 2017, 32, 1104–1106.
- Nicolas, G.; Rovelet-Lecrux, A.; Pottier, C.; Martinaud, O.; Wallon, D.; Vernier, L.; Landemore, G.; Chapon, F.; Prieto-Morin, C.; Tournier-Lasserve, E.; et al. PDGFB partial deletion: A new, rare mechanism causing brain calcification with leukoencephalopathy. J. Mol. Neurosci. 2014, 53, 171–175.
- Biancheri, R.; Severino, M.; Robbiano, A.; Iacomino, M.; Del Sette, M.; Minetti, C.; Cervasio, M.; Del Basso De Caro, M.; Striano, P.; Zara, F. White matter involvement in a family with a novel PDGFB mutation. Neurol. Genet. 2016, 2, e77.
- Tang, L.O.; Hou, B.H.; Zhang, X.N.; Xi, Z.Y.; Li, C.X.; Xu, L. Biallelic XPR1 mutation associated with primary familial brain calcification presenting as paroxysmal kinesigenic dyskinesia with infantile convulsions. Brain Dev. 2021, 43, 331–336.
- Anheim, M.; Lopez-Sanchez, U.; Giovannini, D.; Richard, A.C.; Touhami, J.; N’Guyen, L.; Rudolf, G.; Thibault-Stoll, A.; Frebourg, T.; Hannequin, D.; et al. XPR1 mutations are a rare cause of primary familial brain calcification. J. Neurol. 2016, 263, 1559–1564.
- Lopez-Sanchez, U.; Nicolas, G.; Richard, A.C.; Maltete, D.; Charif, M.; Ayrignac, X.; Goizet, C.; Touhami, J.; Labesse, G.; Battini, J.L.; et al. Characterization of XPR1/SLC53A1 variants located outside of the SPX domain in patients with primary familial brain calcification. Sci. Rep. 2019, 9, 6776.
- Grangeon, L.; Wallon, D.; Charbonnier, C.; Quenez, O.; Richard, A.C.; Rousseau, S.; Budowski, C.; Lebouvier, T.; Corbille, A.G.; Vidailhet, M.; et al. Biallelic MYORG mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 2019, 142, 1573–1586.
- Yang, Q.; Li, J.; Jiao, B.; Weng, L. Primary familial brain calcification in a patient with a novel compound heterozygous mutation in MYORG presenting with an acute ischemic stroke: A case report. Ann. Transl. Med. 2022, 10, 423.
- Gao, L.; Chen, J.; Dong, H.; Li, X. A novel mutation in MYORG leads to primary familial brain calcification and cerebral infarction. Int. J. Neurosci. 2022, 132, 1182–1186.
- Sadok, S.H.; Borges-Medeiros, R.L.; de Oliveira, D.F.; Zatz, M.; de Oliveira, J.R.M. Report of a young patient with brain calcifications with a novel homozygous MYORG variant. Gene 2023, 859, 147213.
- Song, T.; Zhao, Y.; Wen, G.; Du, J.; Xu, Q. A novel MYORG mutation causes primary familial brain calcification with migraine: Case report and literature review. Front. Neurol. 2023, 14, 1110227.
- Chen, Y.; Cen, Z.; Chen, X.; Wang, H.; Chen, S.; Yang, D.; Fu, F.; Wang, L.; Liu, P.; Wu, H.; et al. MYORG Mutation Heterozygosity Is Associated With Brain Calcification. Mov. Disord. 2020, 35, 679–686.
- Chelban, V.; Carecchio, M.; Rea, G.; Bowirrat, A.; Kirmani, S.; Magistrelli, L.; Efthymiou, S.; Schottlaender, L.; Vandrovcova, J.; Salpietro, V.; et al. MYORG-related disease is associated with central pontine calcifications and atypical parkinsonism. Neurol. Genet. 2020, 6, e399.
- Zeng, Y.H.; Lin, B.W.; Su, H.Z.; Guo, X.X.; Li, Y.L.; Lai, L.L.; Chen, W.J.; Zhao, M.; Yao, X.P. Mutation Analysis of MYORG in a Chinese Cohort With Primary Familial Brain Calcification. Front. Genet. 2021, 12, 732389.
- Forouhideh, Y.; Muller, K.; Ruf, W.; Assi, M.; Seker, T.; Tunca, C.; Knehr, A.; Strom, T.M.; Gorges, M.; Schradt, F.; et al. A biallelic mutation links MYORG to autosomal-recessive primary familial brain calcification. Brain 2019, 142, e4.
- Ramos, E.M.; Roca, A.; Chumchim, N.; Dokuru, D.R.; Van Berlo, V.; De Michele, G.; Lieto, M.; Tedeschi, E.; De Michele, G.; Coppola, G. Primary familial brain calcification caused by a novel homozygous MYORG mutation in a consanguineous Italian family. Neurogenetics 2019, 20, 99–102.
- Chen, Y.; Fu, F.; Chen, S.; Cen, Z.; Tang, H.; Huang, J.; Xie, F.; Zheng, X.; Yang, D.; Wang, H.; et al. Evaluation of MYORG mutations as a novel cause of primary familial brain calcification. Mov. Disord. 2019, 34, 291–297.
- Peng, Y.; Wang, P.; Chen, Z.; Jiang, H. A novel mutation in MYORG causes primary familial brain calcification with central neuropathic pain. Clin. Genet. 2019, 95, 433–435.
- Arkadir, D.; Lossos, A.; Rahat, D.; Abu Snineh, M.; Schueler-Furman, O.; Nitschke, S.; Minassian, B.A.; Sadaka, Y.; Lerer, I.; Tabach, Y.; et al. MYORG is associated with recessive primary familial brain calcification. Ann. Clin. Transl. Neurol. 2019, 6, 106–113.
- Fei, B.N.; Su, H.Z.; Yao, X.P.; Ding, J.; Wang, X. Idiopathic basal ganglia calcification associated with new MYORG mutation site: A case report. World J. Clin. Cases 2021, 9, 7169–7174.
- Tekin Orgun, L.; Besen, S.; Sangun, O.; Bisgin, A.; Alkan, O.; Erol, I. First pediatric case with primary familial brain calcification due to a novel variant on the MYORG gene and review of the literature. Brain Dev. 2021, 43, 789–797.
- Chen, S.Y.; Lin, W.C.; Chang, Y.Y.; Lin, T.K.; Lan, M.Y. Brain hypoperfusion and nigrostriatal dopaminergic dysfunction in primary familial brain calcification caused by novel MYORG variants: Case report. BMC Neurol. 2020, 20, 329.
- Kume, K.; Takata, T.; Morino, H.; Matsuda, Y.; Ohsawa, R.; Tada, Y.; Kurashige, T.; Kawakami, H. The first Japanese case of primary familial brain calcification caused by an MYORG variant. J. Hum. Genet. 2020, 65, 917–920.
This entry is offline, you can click here to edit this entry!