Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Limited proteolysis reactions by a disintegrin and metalloproteases (ADAMs) are key events in several paracrine signalling pathways. Therefore, ADAM proteases might also represent master-switches during hepatic fibrosis and other pathophysiologic events. Here, we discuss known and potential fibrosis-associated pathways regulated by ADAM proteases and review the current knowledge on ADAM protease implication in several disease states.
- ADAM
- protease
1. ADAM Proteases
The superfamily of zinc-containing proteases, termed metzincins, is characterised by the presence of an invariant HEXXHXXGXXH zinc-binding motif within the protease domain [5,6]. Metzincins comprise the four subfamilies matrixin, adamalysins, astacins, and bacterial serralysins. The snake venom metalloproteinases (SVMPs), the a disintegrin and metalloproteinases (ADAMs), and ADAMs containing thrombospondin motifs (ADAMTS) build the adamalysin subfamily [7].
The human genome encodes 22 ADAM proteins, of which 10 are considered to be proteolytically inactive [4]. Enzymatically inactive ADAM proteases are thought to be involved in protein folding and protein–protein interactions.
ADAM proteases share an overall domain structure consisting of an N-terminal inhibitory pro-domain, a catalytic metalloprotease domain, followed by a disintegrin domain with a cysteine-rich region, an epidermal growth factor (EGF)-like domain, and finally a transmembrane domain and a cytoplasmic tail (Figure 1a). The family members ADAM10 and 17 are atypical as they are lacking an EGF-like domain. ADAM proteins are synthesised into the endoplasmic reticulum as catalytically inactive proenzymes. Beside its chaperoning function, the N-terminal pro-domain interferes with the catalytic Zn2+-ion and thereby inhibits catalytic activity. Within the Golgi apparatus, proteolytic cleavage, e.g., by the Furin protease, removes the N-terminal pro-domain. For the family members ADAM8 and ADAM28, auto-catalytic removal of the pro-domain was demonstrated [8,9]. Pro-domain removal in ADAM proteases is a prerequisite for full activation of catalytic activity. However, we previously challenged this concept in in vitro experiments using overexpression of Furin-resistent ADAM17 variants. These variants were still able to release tumour necrosis factor α (TNFα) from the cell surface [10]. Interestingly, for ADAM9, 10, and 17 an additional pro-protein convertase cleavage site (upstream site) N-terminal to the previously identified Furin cleavage site (boundary site) was discovered [11]. While cleavage of the C-terminal boundary site by Furin was sufficient for pro-domain removal but dispensable for protease activity, as observed in our experiments, cleavage at the N-terminal upstream site was necessary for the dissociation of the pro-domain and full activation of ADAM proteases [11].
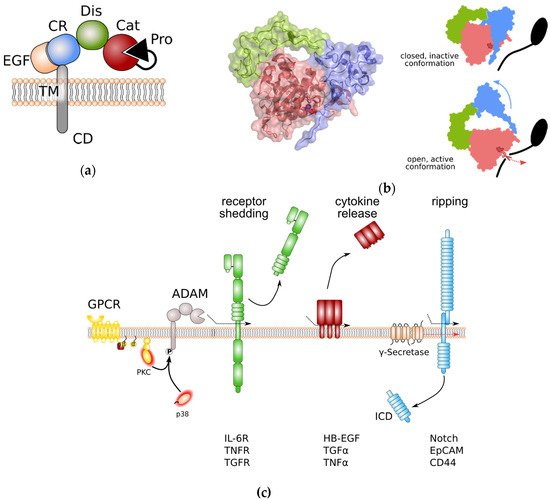
Figure 1. General structure of a disintegrin and metalloprotease (ADAM) proteases and overview of ADAM protease-induced pathways. (a) Domain structure of ADAM proteases. (b) Left panel: Crystal structure of the ADAM10 closed conformation, including the catalytic, disintegrin, and cysteine-rich domain. The Zn2+-binding motif is highlighted in dark red. Colouring of domains is as in (a). Note that the cysteine-rich domain prevents access to the catalytic site. Right panel: Anticipated movement of ADAM10 domains during activation. Colour coding as in (a). The Zn2+-binding motif of the catalytic site is indicated in dark red. (c) General signalling pathways regulated by ADAM proteases. Protein phosphorylation can regulate ADAM protease activity. Limited proteolysis by active ADAM proteases induces removal of receptor ectodomains, release of membrane-bound cytokines and growth factors, and initiates signal transduction by regulated intramembrane proteolysis (RIP). Removal receptor ectodomain can either blunt receptor signalling or induce so-called “trans-signalling” on neighbouring cells. EpCAM: epithelial cell adhesion molecule; EGF: epidermal growth factor; GPCR: G-protein coupled receptor; HB-EGF: heparin-binding EGF-like growth factor; ICD: intracellular domain; IL-6R: interleukin-6 receptor; PKC: protein kinase C; TNFα: tumour necrosis factor α; TNFR: TNFα receptor; TGFα: transforming growth factor α; TGFR: TGFβ receptor.
The catalytic domain of ADAM proteases adopts an overall structure which is common to all metzincins members. The catalytic cleft is positioned between an N-terminal (NSD) and a C-terminal subdomain (CSD) [6,12].
The catalytic domain is anchored by a five-stranded β-sheet which lines the upper side of the catalytic cleft. The lower side of the catalytic cleft is lined by a central α-helix which contains the HExxH motif that supplies two of the histidines coordinating the Zn2+ ion, as well as the glutamate residue that contributes to catalysis. It is followed by a conserved methionine turn that packs against the zinc-binding site [6,12]. Cleavage specificity is determined by the substrate binding pockets. While substrate binding pockets are denoted by S, the substrate residues are denoted by P. Addition of a prime symbol indicates that the position lies in the C-terminal of the cleavage site. For the family members ADAM10 and ADAM17, binding pockets S3, S1, and S1′ determine protease-specific substrate binding. The largest differences between substrate peptides recognized by ADAM10 or ADAM17 are at the P1′ position. While ADAM10 has a deep hydrophobic S1′ pocket and therefore a strong preference for large hydrophobic residues at P1′, the S1′ pocket of ADAM17 is shallower and constrained by an alanine and a valine residue and therefore ADAM17 prefers smaller, non-aromatic hydrophobic residues at P1′ [12].
Lying adjacent to the metalloprotease domain, the disintegrin domain and the cysteine-rich domain seem to be involved in autoregulation. In the absence of substrate binding, these domains fold back to the catalytic centre and therefore limit the access to the substrate specificity pocket of the catalytic site [12,13]. Protein crystallography human ADAM10 has revealed that residues that form the interaction surface between catalytic domain and the cysteine-rich domain are evolutionarily conserved. Furthermore, while overexpression of ADAM10 devoid of its catalytic domain results in dominant negative inhibition of ADAM10 catalytic activity [12], binding of the recently described antibody 8C7 [14] to the ADAM10 cysteine-rich domain releases auto-inhibition and elevates ADAM10 catalytic activity [12]. These data further reinforce the notion that disintegrin and cysteine-rich domains form an auto-inhibitory module forcing ADAM10 into a closed, inactive conformation (Figure 1b). Disintegrin domain and cysteine-rich domain are also involved in binding to substrate proteins. It is speculated that substrate binding releases the auto-inhibitory conformation of disintegrin and cysteine-rich domain (Figure 1b). Interestingly, all human ADAMs contain a hyper-variable region within the cysteine-rich domain with low sequence homology which may be critical for substrate binding and might contribute to the substrate selectivity of ADAM family members [13]. The fact that binding of the endogenous inhibitor tissue inhibitor of metalloproteases 3 (TIMP3) to the catalytic site of the recombinant isolated catalytic domain of ADAM17 is faster in the absence of disintegrin domain and cysteine-rich domain [15] supports the hypothesis that ADAM17 has a similar auto-inhibitory mechanism as ADAM10.
Protease activity of ADAMs can either be constitutive or activated by extracellular and intracellular signalling pathways. Members of the membrane multi-pass protein families inactive rhomboids (iRhoms) [16,17] and tetraspanins (Tspans) [18,19] have been demonstrated to promote maturation of ADAMs on the secretory pathway and to be involved in the activation process via multiple phosphorylation events.
The rhomboids are a family of evolutionarily conserved multi-transmembrane proteins. The non-protease rhomboids lack amino-acid residues that are essential for protease activity. In particular the members iRhom1 and iRhom2 were identified to interact with ADAM17 as early as in the endoplasmic reticulum in order to promote trafficking to the Golgi [16,17]. iRhom2 was demonstrated to be essential for ADAM17 activities such as TNFα release, particularly in myeloid cells [16,17]. iRhoms are furthermore discussed to regulate substrate selectivity of ADAM17 [20].
Tspans are a family of small and evolutionary conserved proteins that span the membrane four times, resulting in the formation of a short and a large extracellular loop. The latter is involved in particular in client protein interaction. Tspans have the ability to form large interacting networks, the so-called tetraspanin-enriched membrane microdomains (TEM) which represent a signalling platform similar to lipid rafts [21]. Previously, it was demonstrated that in particular members of the TspanC8 family, that is characterised by the presence of eight cysteine residues within the large extracellular loop, bind to ADAM10 within the endoplasmic reticulum and promote its trafficking to the plasma membrane [18,19].
The cytoplasmic tail of ADAM proteases is discussed to regulate catalytic activity of some family members (Figure 1c). It contains several sites that can be phosphorylated by diverse cytoplasmic kinases such as protein kinase C (PKC) [22,23], extracellular regulated kinase (ERK) [24,25], and p38 [26]. In particular, engagement of G-protein coupled receptors (GPCRs) that are coupled to an αq/11 subunit results in the downstream activation of phospholipase C (PLC), an increase in the second messengers diacylglycerol (DAG), inositol triphosphate (IP3), and Ca2+, and subsequent activation of PKCs [27,28,29]. While activation of ADAMs by PKCs is less well understood, activation of ADAM17 through C-terminal phosphorylation by ERK and p38 was shown to disrupt ADAM17 dimerization and thereby to release the inhibitory binding of tissue inhibitor of metalloproteases (TIMP) 3 [30].
Recently, basic residues within the membrane-proximal cysteine-rich domain of ADAM17 have been claimed to confer binding to phosphatidylserine (PS) [31]. Only under conditions of cell death, such as apoptosis and necroptosis, is PS exposed to the outer leaflet of the plasma membrane. Under apoptotic conditions, PS exposure and binding to ADAM17 cysteine-rich membrane proximal domain (MPD) alter ADAM17 conformation, releasing autoinhibition and inducing cleavage of substrate molecules [31,32]. The MPD of ADAM17 has been discussed to undergo an isomerization of disulfide bridges and therefore to either adopt an “open” or a “closed” conformation [33]. PS binding to isolated recombinant MPD only occurred when MPD was present in an “open” conformation [31], suggesting that ADAM17 activation is regulated at two levels. Interestingly, PS-binding seems to be a prerequisite for ADAM17 activation in general. Therefore, transient PS exposure might not only be restricted to apoptotic conditions.
The C-terminus of ADAM proteases is furthermore important for their negative regulation via endocytosis. Binding of the cytosolic adaptor proteins Grb2 to ADAM12 and AP2 to ADAM10 was demonstrated to mediate internalisation via the clathrin-dependent pathway [34,35]. Interestingly, overexpression of dominant negative dynamin K44A prevented internalisation but enhanced proteolytic processing of surface-bound ADAM10 [36]. ADAM17 was also shown to be internalised upon phorbol-12-myristate-13-acetate (PMA)-stimulation [37]. Complex formation of ADAM17 with iRhoms and the FERM domain-containing protein (FRMD) 8 at the cell surface was identified to prevent ADAM17 internalisation and lysosomal degradation [38,39]. Once internalised, interaction of ADAM17 with the sorting protein phosphofurin acidic cluster sorting protein (PACS) 2 at early endosomes promotes recycling of ADAM17 back to the cell surface [40].
There is some evidence that in the liver, ADAM protease activity is regulated by bile acids. Ursodeoxycholic acid was shown to inhibit PMA-induced ADAM17 activity on both the HepG2 hepatocellular cell line and the hepatic stellate cell line LX-2 [41]. In contrast, deoxycholic acid (DCA) enhanced ADAM17 catalytic activity and release of amphiregulin in colorectal and pancreatic cancer cell lines. This effect was mediated via binding of DCA to the G protein-coupled bile acid receptor 1, also termed TGR5, and subsequent c-Src activation [42].
2. Regulation of Liver Epithelial Cell Regeneration
Epithelial cell (i.e., hepatocyte or cholangiocyte) damage is common to almost all liver insults. Hepatocyte apoptotic bodies and damage-associated patterns (DAMPs) lead to rapid activation of Kupffer cells (KCs), a liver-resident macrophage population. In these cells, the proteolytic release of membrane-bound pro-inflammatory cytokines such as TNFα and proliferative signals such as cleavage of membrane-bound epidermal growth factor receptor (EGFR) ligands are most likely orchestrated by ADAM17. TNF-release from myeloid cells via ADAM17 can be induced the bacterial cell wall component lipopolysaccharide (LPS) [43,44] through toll-like receptor (TLR) 4 engagement [45]. During chronic liver disease, increased translocation of gut microbial products is enhanced and TLRs on KCs activated via pathogen-associated molecular patterns (PAMPs). Identification of TLR4 single-nucleotide polymorphisms [46] and use of TLR4-deficient mice [47] have demonstrated the importance of TLR4 for the development of fibrotic disease. While experimental proof is still lacking, it is likely that myeloid ADAM17 is involved in the development of inflammation and cancer in liver, lung and the intestine.
HSCs release EGFR ligands to promote hepatocyte proliferation [3]. Autocrine EGFR activation on KCs was recently identified to be critical for the induction of interleukin-6 (IL-6) release in a murine HCC model [48]. It is therefore conceivable that EGFR ligand release from HSCs induces IL-6 release from myeloid cells. On target cells, IL-6 binds to the membrane-bound IL-6 receptor (IL-6R) α, before engaging a gp130 homodimer that transduces the signal into the cell. This process is called “IL-6 classic signalling”. The proteolytic release of the soluble IL-6 receptor (sIL-6R) from KCs and infiltrated myeloid cells which is most likely mediated by ADAM17, in conjunction with secreted IL-6 creates a strong regenerative signal for hepatocytes [49,50]. On target cells, the complex of IL-6/sIL-6R can bind and therefore activate gp130, even on cells that do not express the membrane-bound IL-6R. This process is termed “IL-6 trans-signalling” [51]. Persistent activation of IL-6 trans-signalling on damaged hepatocytes can ultimately lead to the development of hepatocellular carcinoma (HCC) [52]. On the other hand, it has been shown that ectodomain shedding of c-Met is mediated by ADAM10 [53]. While experimental evidence is still lacking, it is conceivable that the overwhelming hepatocyte growth factor (HGF)-mediated mitogenic response of hepatocytes is dampened by ADAM10. This is supported by the fact that genetic c-Met-deficiency accelerates fibrosis in a CCl4-model [54]. ADAM10 is essential for hepatocyte homeostasis. Consequently, loss of hepatic ADAM10 resulted in spontaneous development of liver fibrosis [55]. This may, at least in part, be due to an accumulation of liver progenitor cells [55] that have previously been linked to activation of HSCs [56,57]. Furthermore, release of TNFα, presumably via ADAM17, sustains inflammation and promotes hepatocyte cell death [58].
Through the secretion of chemokine (C-C motif) ligand (CCL)2, activated KCs promote the CCR2-dependent recruitment of a pro-fibrotic CD11b+F4/80+Ly6Chi macrophage population. ADAM protease-mediated release of soluble IL-6 receptor (sIL-6R) and induction of IL-6 trans-signalling furthermore induces migration of KCs/myeloid cells to the site of liver damage [52]. Secretion of pro-inflammatory and pro-fibrotic mediators by infiltrated myeloid cells promotes activation of HSCs [2]. Experimental depletion of CD11b+ cells in murine models of liver fibrosis has demonstrated that monocyte recruitment is essential for liver fibrosis [59,60].
Activation of HSCs and trans-differentiation into myofibroblasts is a key event during fibrosis of the liver. It is now a well-accepted concept that HSC activation and fate can be divided into (1) initiation, (2) perpetuation, and (3) clearance. During initiation, HSCs are rendered responsive to many extracellular signals e.g., through rapid up-regulation of receptor molecules. Perpetuation refers to events that sustain and amplify HSC activation. During clearance activated HSCs are removed either through induction of HSC apoptosis or the reversal into an inactive, quiescent state [3].
Transforming growth factor (TGF) β secreted from pro-fibrotic macrophages is the strongest inducer of HSC activation. Engulfment of epithelial cell apoptotic bodies by HSCs or DAMPs also contributes to HSC activation [61]. Rapidly after initial activation, HSCs up-regulate platelet-derived growth factor (PDGF) and its corresponding PDGF receptor β. PDGF is a strong HSC mitogen and promotes HSC proliferation [3].
Importantly, ADAM17 has recently been shonw to be involved in several severe disease states such as pulmonary emphysema (PMID: 33181031), pancreatitis (PMID: 36215509), tobacco smoke carcinogen-induced lung tumorigenesis (PMID: 31257400), KRAS-addicted lung cancer (PMID: 30833304), kidney fibrosis (PMID: 27642633), colon cancer (PMID: 29472497) and metastasis (PMID: 34919140). Therapeutic blockade of ADAM17 activity with an engineered recombinant pro-domain of ADAM17 has been successfully applied in various animal models of human diseases (PMID: 27642633, PMID: 34919140, PMID: 30833304) underlining the importance of this protease and pointing to novel therapeutic principles in these diseases.
This entry is adapted from the peer-reviewed paper 10.3390/cells8101226
This entry is offline, you can click here to edit this entry!