1. Lysosomes Enable Drug Release from GNM-Containing Nanocarriers, Whereupon Drugs Escape from Lysosomes
Since most chemotherapeutic agents achieve their anticancer effects by acting on cell organelles other than lysosomes (e.g., the cell nucleus, mitochondria, and endoplasmic reticulum), they must escape from the endosome/lysosome to exert their effect and avoid degradation by acid hydrolases [
54]. Recently, many novel polymers, pH-sensitive peptides, proteins, and other endosomolytic agents have been synthesized to facilitate the endosomal escape [
55]. Several mechanisms that are responsible for the lysosomal escape of nanoparticles could also apply to graphene-based DDSs (
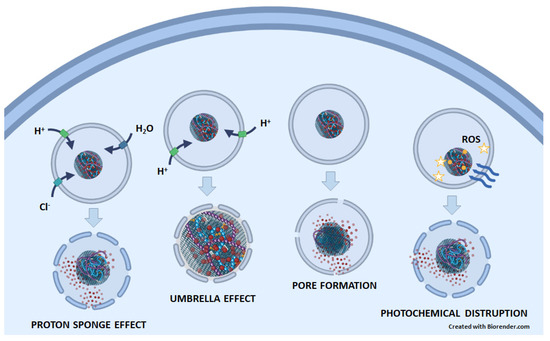
Figure 3). (1) The proton sponge effect is the ability of molecules, such as polyethylenimine (PEI) and polyamidoamine (PAMAM) dendrimers, that contain amine groups with buffering capacity to absorb protons, which prevents the acidification of endosomes/lysosomes. To achieve the desired pH, ATPase continues to pump protons into the endosome/lysosome, causing an influx of chloride ions and water osmosis. This process leads to an increase in pressure within the lysosomes and ultimately results in a lysosomal burst. (2) The umbrella effect is an extension of the proton sponge effect. When amines are protonated, charge repulsions cause the structure of nanoparticles to expand, leading to the rupture of endosomes/lysosomes. (3) Pore formation: Nanoparticles can fuse directly with the membrane of endosomes/lysosomes, inducing membrane stress and internal membrane tension, which can lead to the formation of pores. (4) Photochemical disruption is a mechanism in which a photosensitizer, when exposed to light, generates ROS that can destroy the integrity of the endosomal/lysosomal membrane [
55,
56,
57].
Figure 3. The possible mechanisms of endosomal/lysosomal escape of anticancer drugs (red circles) released from GNMs. In the proton sponge effect, GNM-bound molecules containing amine groups with buffering capacity absorb protons. Therefore, the ATPase continuously pumps protons into the endosome/lysosome, which is followed by an influx of chloride ions, water osmosis, increased pressure, and, finally, lysosomal rupture. In the umbrella effect, protonation of amines leads also to charge repulsion and nanoparticle expansion, which causes endosomal/lysosomal rupture. By inducing membrane stress and internal membrane tension, nanoparticles can lead to the formation of pores on the endosome/lysosome membrane. In photochemical disruption, a light-irradiated (blue wavy arrows) photosensitizer (yellow circles) attached to a GNM-containing nanocarrier generates ROS (yellow stars), which destroys the integrity of the endosomal/lysosomal membrane.
For an anti-cancer effect, it is not necessary for the entire graphene-based DDS to leave the endosome/lysosome, only the anti-cancer drug. To achieve this, the drugs must first be separated from the GNM-containing nanocarrier. This can happen in several ways. (1) The low pH environment within lysosomes triggers the protonation of graphene-based DDSs, leading to electrostatic repulsion between positively charged drugs and nanocarriers [
39,
40]. (2) Low pH triggers the breakdown of the π–π interaction, the supramolecular forces that arise between unsaturated (poly)cyclic molecules with π-orbitals [
58]. This has been observed for the π–π interaction between GNMs and doxorubicin [
59,
60,
61], proflavine [
61], or Chlorin e6 [
62]. (3) The breakdown of chemical bonds. For example, the diselenide bond between GO and PAMAM was broken by a low lysosomal pH [
15] or a high lysosomal ROS [
13], resulting in the release of the drug. Next, the studies describing how anticancer drugs are released from GNM-containing nanocarriers and the mechanisms of their endosomal/lysosomal escape were listed (
Table 1).
Table 1. Lysosomes enable drug release from GNMs, which is followed by endosomal/lysosomal escape of the drug. → denotes causal connection; ↑ denotes increase; ? denotes unknown mechanism. Abbreviations for graphene-based DDSs in order of appearance: GPCP/miR-21i/ICG, poly(l-lysine)-modified graphene conjugated with citraconine and polyamidoamine loaded with indocyanine green and miR-21i; ICG/GPP, graphene oxide modified with poloxamer 188-modified polyamidoamine-dendrimer; GO/AuNS-PEG/Ce6, PEGylated graphene/gold nanostar hybrid loaded with Chlorin e6; FPS, fluorinated GO modified with PEI and sericin; FPS-Cur, FPS loaded with curcumin; ICG/DOX/GO-PPF68, graphene oxide conjugated with polyamidoamine-pluronic F68 loaded with doxorubicin and indocyanine green; DOX@-GO@AuNR, gold nanorods wrapped in graphene oxide and loaded with doxorubicin; Ag-GO/DOX, graphene oxide loaded with doxorubicin and Ag; NGO/DOX@SPC-FA, nanographene oxide loaded with doxorubicin, co-coated with soy phosphatidylcholine and modified with a PEGylated folic acid; NGO-PEG-DA/DOX, nanographene oxide conjugated with polyethylene glycol and poly(allylamine hydrochloride), modified with 2,3-dimethylmaleic anhydride and loaded with doxorubicin; PK5E7(PEI-rGO), poly(ethylene imine) coated with poly(ethylene imine)-poly(l-lysine)-poly(l-glutamic acid) and conjugated with reduced graphene oxide; GPC-NP, graphene oxide transformed into 3D in the presence of cisplatin and loaded with proflavin; GDC-NP, graphene oxide transformed into 3D in the presence of cisplatin and loaded with doxorubicin; GT/IR820/DP-CpG, 1,2-distearoyl-3-phosphatidylethanolamine and polyethylene glycol conjugated with alkyl triphenylphosphonium and graphene oxide, loaded with diphenylmethylsilane-substituted CPG and photosensitizer IR820.
Wu and colleagues constructed the smart vehicle GPCP, which is composed of PLL-modified graphene conjugated with citraconine (Cit) as a charge-convertible inner layer, and PAMAM dendrimer as a cationic outer layer. GPCP was loaded with photosensitizer indocyanine green (ICG), which generates heat and ROS when irradiated in the near-infrared (NIR) and miR-21i, an oligonucleotide inhibitor of oncogenic microRNA 21. After endocytosis of the GPCP/miR-21i/ICG by MDA-MB-231 triple-negative breast cancer cells, both ICG and miR-21i were released at the acidic pH of lysosomes due to the acid-triggered charge reversal effect. Furthermore, charge-reversed GPCP destabilized the endosomal membrane through the proton sponge effect, allowing both ICG and miR-21i to escape from the endosomes/lysosomes and exert their anticancer effects. Importantly, the complex demonstrated potent antitumor activity in the NIR-irradiated MDA-MB-231 mouse model [
12].
Wang and colleagues modified GO with a Poloxamer 188-modified PAMAM dendrimer, which exhibits a strong proton sponge effect. The resulting GPP complex was then loaded with the ICG. The ICG/GPP entered MCF-7 breast cancer cells by endocytosis. ROS induced the breakage of diselenide bonds between GO and PAMAM-Poloxamer 188, resulting in the release of ICG. The proton sponge effect combined with oxidative stress led to the destabilization of the lysosomal membrane, allowing for the lysosomal escape of the drug. Finally, upon NIR irradiation, GO produced heat while ICG produced both heat and ROS, leading to tumor cell death [
13].
Theranostics is a term used to describe the simultaneous diagnosis and treatment of a disease. In a study by Wu et al., a graphene/gold nanostar hybrid was stabilized by PEGylation in a GO/AuNS-PEG complex. PEGylation is a widely used method to improve the hydrophilicity and biocompatibility of nanocomplexes [
65]. The photosensitizer Chlorin e6 (Ce6) was loaded onto the GO/AuNS-PEG complex through π–π stacking. The resulting phototheranostic GO/AuNS-PEG/Ce6 was endocytosed into the lysosomes of EMT6 mouse breast cancer cells. Upon NIR irradiation, the lysosomal membrane was ruptured by the heat and ROS generated by GO/AuNS-PEG and Ce6, respectively. This facilitated the release of the complex from the lysosomes and its targeting of the mitochondria, ultimately inducing cell death. Treatment of EMT6 tumor-bearing mice with GO/AuNS-PEG/Ce6 and NIR irradiation enabled in-vivo photothermal imaging by an infrared camera and strongly reduced tumor growth [
62].
Jahanshahi and co-workers modified fluorinated GO with two molecules with pH-dependent charge reversal properties, PEI and sericin from silkworm, to form nanocarriers named FPS. FPS was loaded with the hydrophobic natural antitumor drug curcumin. Formed FPS-Cur contained two different pH-sensitive amide linkages that were negatively charged at blood pH (≈7.4), resulting in a prolonged circulation time. At mildly acidic conditions (pH ≈ 6.5 for tumor extracellular matrix and pH ≈ 5.5 for endosomes/lysosomes), the amide linkages in FPS-Cur underwent hydrolysis, causing the complex to switch its charge from negative to positive. The induced electrostatic repulsion led to the opening of the complex’s structure, allowing for the release of curcumin. Curcumin then entered the nucleus, triggering apoptosis and necrosis of HeLa cervical cancer cells [
14].
The hydrophobic antitumor drug and DNA synthesis inhibitor doxorubicin (DOX) binds to graphene and its derivatives through both hydrophobic and π–π stacking interactions. Wang et al. conjugated GO with PAMAM-Pluronic F68 (PPF68) via a diselenide bond and loaded it with DOX and ICG. The formed ICG/DOX/GO-PPF68 was internalized into multidrug-resistant (MDR) MCF-7/ADR human breast cancer cells by endocytosis, where PPF68 protected the drugs from leakage and degradation. ROS, which was generated by NIR-irradiated ICG, triggered the cleavage of the diselenide bond in the complex, resulting in the release of DOX. The proton sponge effect of PAMAM and ROS induced LMP and allowed the lysosomal escape of DOX. Upon NIR irradiation, the ROS and heat generated by ICG, the heat generated by GO, and the antiproliferative effect of DOX decreased the viability of tumor cells. Notably, the intravenously injected composite accumulated in the tumor tissues of MCF-7/ADR tumor-bearing mice due to the EPR effect and greatly reduced the tumor growth of the animals upon NIR irradiation [
15].
Deng et al. loaded DOX on GO-wrapped gold nanorods, which enabled complex detection by Surface-enhanced Raman spectroscopy (SERS). At the beginning of treatment with DOX@-GO@AuNR complex, the SERS signals from GO and DOX overlapped near HeLa cells. Later, the π–π DOX/GO interaction was broken at low lysosomal pH, as evidenced by a decrease in DOX signals compared to GO SERS signals, indicating the appropriate time to irradiate the cells. The photothermal effect of NIR-irradiated GO and antiproliferative effects DOX reduced cell viability. The antitumor effect of DOX@-GO@AuNR was confirmed in NIR-irradiated HeLa-bearing mice [
63].
Huang et al. developed a GO-based nanocarrier and loaded it with DOX and Ag nanoparticles as SERS substrates. The Ag-GO/DOX complex was endocytosed by Ca Ski human cervical carcinoma cells, and the low lysosomal pH disrupted the π–π interaction between DOX and GO surface, resulting in the release of DOX. SERS tracking confirmed that DOX escaped from the lysosomes into the cytoplasm and entered the nucleus, while GO remained in the cytoplasm [
59].
Ma and colleagues utilized both π–π stacking and hydrophobic interactions to load DOX onto nanographene oxide (NGO). They then co-coated the formed hydrophobic NGO/DOX with a biomimetic membrane made of soy phosphatidylcholine (SPC) and modified it with a PEGylated lipid-FA (where FA stands for folic acid). The resulting NGO/DOX@SPC-FA complex demonstrated long-term blood circulation in HeLa cervical tumor-bearing mice and preferential entry into the tumor tissue due to the EPR effect. The FA moiety facilitated the binding of the complex to the FA receptor on cancer cells, followed by clathrin-dependent endocytosis. DOX was released from NGO in the acidic lysosomal environment due to the protonation effect, escaped from the lysosomes, and entered the nucleus to exert its anticancer effect [
60].
Feng et al. prepared a complex of NGO, PEG, positively charged poly(allylamine hydrochloride), and 2,3-dimethylmaleic anhydride (DA) with pH-dependent charge reversibility, and then loaded it with DOX. The formed NGO-PEG-DA/DOX was negatively charged at a physiological pH of 7.4, whereas it was positively charged in the tumor microenvironment (pH = 6.8) due to the cleavage of DA. This enhanced its uptake in MCF-7 cells and provided tumor-specific cytotoxicity. At a lysosomal pH of 5, DOX was released from the complex due to its protonation-induced hydrophilicity/solubility. Moreover, electrostatic repulsion between the protonated DOX and positively charged (charge-reversed in low pH) NGO-PEG-DA also contributed to DOX release. The NGO-PEG-DA/DOX complex exhibited a synergistic anticancer effect of DOX-dependent chemotherapy and NIR-irradiated NGO-dependent photothermal therapy [
40].
Ryu and colleagues conjugated poly(ethylene imine)-poly(l-lysine)-poly(l-glutamic acid) (PKE) with charge-conversion properties, a proton sponge polymer PEI, and rGO. The resulting PK5E7(PEI-rGO)/DOX complex was negatively charged at pH 7.4 and positively charged at pH 6. The DOX-loaded complex rapidly released the anticancer drug under lysosomal conditions due to electrostatic repulsion between the positively charged DOX and PEI-rGO. PK5E7(PEI-rGO) showed potent anticancer activity in HeLa and A549 lung adenocarcinoma cells under mildly acidic conditions mimicking the tumor microenvironment [
39].
In the presence of the anticancer drug cisplatin, Nandi et al. transformed 2D sheets of GO, bound to the DNA-damaging drugs proflavin or DOX by π–π interaction, into 3D spherical GPC-NP and GDC-NP complexes, respectively. After clathrin-mediated endocytosis in HeLa cells, proflavin and DOX were released from the complexes under the influence of lysosomal acidity. Carboxylate bonds were broken, and cisplatin was also released from the complexes at low pH, returning the 3D GO to its 2D form. Ultimately, the cells died by apoptosis [
61].
Wu et al. developed a mitochondria-targeting carrier (GT) by attaching the amphiphilic polymer DSPE-PEG (composed of 1,2-distearoyl-3-phosphatidylethanolamine (DSPE) as the hydrophobic tail and PEG as the hydrophilic chain) to the mitochondria-targeting moiety alkyl triphenylphosphonium (TPP) and GO. The resulting GT was then combined with the immunoconjugate diphenylmethylsilane-substituted CPG (DP-CpG) and the photosensitizer IR820. The formed GT/IR820/DP-CpG complex destabilized lysosomal membrane, escaped lysosomes, and targeted mitochondria of EMT6 mouse breast cancer cells. Upon NIR irradiation, the complex generated heat and ROS, leading to apoptosis. In EMT6 tumor-bearing mice, GT/IR820/DP-CpG produced PDT- and PTT-mediated anticancer effects and activated an anticancer immune response [
64].
The ability of acidic lysosomal milieu to induce physiochemical changes in nanocomplexes was mainly used for that purpose. With the exception of positively charged ICG/DOX/GO-PPF68 [
13], all other complexes were negatively charged but became positive after protonation inside the lysosomes, which induced charge repulsion and separation of the drugs from the rest of the complexes [
12,
14,
39,
40,
60]. While the ability of low lysosomal pH to provoke break downs of π–π interactions [
59], amide [
14], and carboxylate [
61] bonds, as well as the ability ROS produced by NIR-irradiated photosensitizer to disrupt diselenide bonds, was exploited, there has been no study that clearly utilized the presence of hydrolytic enzymes for this purpose. Mechanisms of lysosomal drug escape have only been superficially addressed. While the drug escape through the lysosomal membrane, which has been damaged by the heat generated by NIR-irradiated GNMs or the combined effects of heat and ROS produced by NIR-irradiated photosensitizers, has been clearly established, [
62,
64], the proton sponge mechanism was more hypothesized due to the presence of the ensosomolytic molecule PAMAM than proven [
12,
13,
15]. Moreover, lysosomal escape was stated, but its mechanism was not investigated in the rest of the above-mentioned studies [
14,
39,
40,
59,
60,
61]. Although the fact that antitumor drugs escaped from lysosomes seems to be more important for their antitumor activities than how they escaped from lysosomes, an elucidation of the mechanisms of escape would be a useful guide for future synthesis of graphene-based DDSs and should, therefore, be clearly elucidated.
5.2. GNMs Induce Lysosomal Cell Death
Lysosomal escape may be associated with LMP. The consequence of LMP is the leakage of lysosomal enzymes into the cytoplasm. Cathepsins B, D, and L retain their activity at neutral pH, degrading cytoplasmic proteins and inducing LCD. The type of LCD induced depends on the severity of lysosomal damage; extensive LMP, with leakage of cathepsins and protons into the cytoplasm, triggers necrosis, while limited LMP triggers apoptosis. Additionally, LMP may also play a role in the initiation of autophagic cell death, necroptosis, and ferroptosis [
3].
LCD may occur subsequent to the escape of nanoparticles from lysosomes via various mechanisms, including the proton sponge effect, the umbrella effect, pore formation, or photochemical disruption of the lysosomal membrane [
56,
66,
67]. However, in some cases, LCD may not occur due to the dominance of other mechanisms of cell death. For instance, a study involving the treatment of tumor cells with ICG/DOX/GO-PPF68 detected clear LMP, but the authors attributed the antitumor effects to the antiproliferative activity of DOX, ROS generated by NIR-irradiated ICG, and hyperthermia caused by NIR-irradiated GO and ICG [
15]. Furthermore, in another study, GO nanosheet-induced LMP led to lysosomal alkalinization, which inhibited autophagic degradation and, ultimately, triggered apoptosis, but not LCD [
10].
It is crucial to distinguish between endosomal and lysosomal escape as endosomes have a lower acidity level and hydrolytic enzymes content. Consequently, LCD is more likely if the drug escapes from mature lysosomes rather than endosomes [
56]. It is important to note that escape of anticancer drugs through pore formation may not necessarily result in the leakage of cathepsins. The formed pores may be large enough to allow for the release of the drug but not the hydrolytic enzymes. For example, Cathepsin B has a molecular weight of approximately 25,000 Da [
68], while the molecular weight of DOX is only 544 Da [
69].
Oxidative stress is one of the main LMP inducers [
3]. Accelerated metabolism of tumor cells results in increased breakdown of iron-containing proteins and iron accumulation in lysosomes. This iron can convert ROS into highly reactive OH• via the Fenton reaction [
70], which in turn disrupts lysosomal membrane integrity. Despite its short half-life, OH• is a very toxic molecule due to its high reactivity. OH• causes breaks in DNA strands, modifies DNA bases, and disrupts protein structure and function. OH• also triggers the peroxidation of lipids, which are essential components of cell membranes, including lysosomal membranes [
71]. Lysosomes lack the antioxidant enzymes superoxide dismutase, catalase, and glutathione peroxidase [
3], making them particularly vulnerable to oxidative stress. Accordingly, oxidative stress has been identified as the mediator of LMP in all studies involving both GNMs and LMP (
Table 2).
Table 2. GNMs and graphene-based DDSs induce LMP. → denotes causal connection; ↓ denotes decrease/inhibition. ROS—reactive oxygen species. Abbreviations for GNMs/graphen-based DDSs in order of appearance: GNSs, graphene nanosheets; DSPE-PEG2000-FA, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethylene glycol)-2000]; Ce6, Chlorin e6; GO, graphene oxide; rGO-Ru-PEG, reduced graphene oxide loaded with Ru(II)-polypyridyl complex modified with polyethylene glycol; DHA, dihydroartemisinin; Tf, transferrin.
In a study conducted by Liu et al., rat basophilic leukemia RBL2H3 cells endocytosed graphene nanosheets (GNSs), which, due to their sharp edges and rough surface, damaged the lysosomal membrane, thereby inducing LMP. GNSs also disrupted the mitochondrial electron transport chain, leading to excessive ROS production. This oxidative stress further stimulated LMP and mitochondrial membrane depolarization, ultimately resulting in apoptotic cell death [
67].
Tian et al. developed a complex by assembling 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethylene glycol)-2000] (DSPE-PEG2000-FA) and a photosensitizer-labeled peptide (Ce6-Pep) on the surface of GO. This complex was internalized by HeLa cells via folate receptor-mediated endocytosis and localized in lysosomes. In lysosomes, cathepsin B cleaved the peptide, releasing Ce6 from GO. Upon red light irradiation, Ce6 induced the formation of singlet oxygen
1O
2, which triggered the release of cathepsin B from lysosomes to cytosol and led to LCD [
16].
Zhang and colleagues attached a PEG-modified Ru(II)-polypyridyl complex to rGO through π–π stacking and hydrophobic interactions. Upon internalization, the rGO-Ru-PEG complex was detected in lysosomes of A549 cells. Low pH and the heat produced by NIR irradiated-rGO caused the release of Ru-PEG from the rGO surface. The irradiation of cells with NIR for PTT and blue light for PDT resulted in the synergistic induction of apoptosis through the generation of ROS and the release of cathepsin B from lysosomes into the cytosol. Notably, treatment with rGO-Ru-PEG and irradiation with both NIR and blue light significantly reduced tumor growth in A549 tumor-bearing mice [
17].
Liu et al. developed DHA-GO-Tf by combining the anti-malarial drug dihydroartemisinin (DHA), nanoscale GO, and transferrin (Tf), an iron transporter that can target tumor cells expressing Tf receptors. DHA-GO-Tf was endocytosed in EMT6 cells. In the acidic environment of lysosomes, iron (Fe
+3) was released from Tf and reduced to Fe
+2 by lysosomal ferrireductase. The reaction between DHA and Fe
2+ generated ROS, leading to oxidative damage of lysosomes and cytotoxicity. DHA-GO-Tf was found to preferentially accumulate in the tumor tissue of EMT6-bearing mice and induce complete tumor regression [
33].
In above-mentioned studies, LMP appears to be only one of the steps leading to tumor cell death. Considering the huge instability of cancer lysosomes, the ability of using LCD in GNM-based tumor therapy should be investigated in more detail. It is useful to determine whether there is a relationship between the size and charge of GNMs and their capacity to destabilize lysosomes. The ability of GNMs to induce LMP could be stimulated by their modification with amine-modified polystyrene, as this has been shown to be successful with other nanoparticles [
72]. In addition, loading GNM with anticancer drugs that can themselves induce LMP, such as vincristine, vinblastine, paclitaxel, cisplatin, and doxorubicin [
73], could be used for this purpose.
3. GNMs Induce Tumor Cell Death by Suppressing (Auto)Lysosomal Degradation
In the process of autophagy, exhausted/damaged intracellular macromolecules and organelles are entrapped in double-membrane autophagosomes. Autophagosomes then fuse with lysosomes in autolysosomes where sequestered content is degraded by acid hydrolases. Since autophagy is primarily responsible for degrading intracellular cytoplasmic content, it is unclear how externally added GNMs are included in autophagic vacuoles [
11].
On the other hand, several studies have shown that GNMs can induce tumor cell death by suppressing autophagic degradation. In the research conducted by Xiaoli and co-workers, GO induced oxidative stress, mitochondrial damage, and reduced energy production, thereby stimulating incomplete autophagy in human neuroblastoma cells SH-SY5Y. The non-degradable GO accumulated in lysosomes, causing steric hindrance that elevated lysosomal pH and suppressed the degradative capacity of lysosomal enzymes such as acid phosphatase and cathepsin B. This resulted in the accumulation of dysfunctional mitochondria and ultimately contributed to mitochondria-dependent apoptosis [
9]. Feng et al. demonstrated that GO nanosheets induced LMP, impaired the acidity of lysosomes, and thus suppressed the activity of cathepsin B and acid phosphatase in PC12 rat pheochromocytoma-derived cells. The impairment of lysosomal degradation led to abnormal accumulation of the autophagic proteolysis substrate p62, which contributed in part to the induction of apoptosis [
10]. Similarly, Zhang and coworkers demonstrated that GO induced lysosomal alkalization, inhibited cathepsin B, and induced p62 accumulation and apoptosis in F98 rat astroglioma cells [
11]. After internalization in A549 cells via clathrin-mediated endocytosis, GO-chloroquine nanoconjugate (GO-Chl) was found to stimulate the synthesis of autophagosomes but suppress their fusion with lysosomes. This resulted in the blockade of the autophagy flux and activation of necroptotic death, as demonstrated by Arya et al. [
53].
Conversely, through the selective degradation of mitochondria and cell survival proteins, prolonged and overactivated autophagy can cause apoptotic, necrotic, or necroptotic death of tumor cells, or itself be an alternative mechanism of cell death (programmed cell death Type II). Consistent with the cytotoxic role of autophagy, autophagy was induced by the combined treatment with GO and the cisplatin-stimulated necrosis of colorectal carcinoma cells [
80], whereas autophagy induced by photoactivated GQDs stimulated apoptosis in glioma cells [
78].
Therefore, either incomplete cytoprotective autophagy [
9,
10,
11,
53] or autophagy that is itself cytotoxic [
78,
80] could be useful in cancer therapy with GNMs. Moreover, it might be valuable to combine widely used chemotherapeutic agents that induce pro-survival autophagy with GNMs that block autophagy flux [
9,
10,
11,
53]. On the other hand, complete pro-survival autophagy induced by GNMs [
76,
77,
79] could be suppressed by drugs such as anti-malaric chloroquine, or anticancer drugs that have been shown to suppress autophagy flux.
GNMs were also shown to suppress lysosomal enzymes unrelated to autophagy. Yang et al. showed that PEG–GO nanocarriers increased the lysosomal accumulation of the anticancer drug tamoxifen in human hepatoma HepG2 cells. The PEG-GO/tamoxifen complex inhibited the lysosomal enzyme phospholipase, resulting in a lysosomal storage disorder called phospholipidosis [
82]. This condition may be cytotoxic [
83], but the sequestration of tamoxifen in lysosomes is expected to prevent its antitumor effects mediated by a blockade of estrogen receptors [
82]. However, the effect of nanocarriers on the anticancer activity of the drug still requires investigation.
4. Lysosomes Enable Detection of Cancer Cells by the GNMs
Finally, lysosomes are not only directly involved in the antitumor activity of GNMs, but the specificities of the lysosomal environment, such as its acidity or the presence of proteolytic enzymes, may also facilitate the identification of tumor cells, as demonstrated in the following studies.
Mosaiab and colleagues loaded DOX onto a (CA-BDP)-PPDN/rGO complex, composed of chloro-3′,4′-dihydroxyacetophenone (CA), a fluorescent dye boron-dipyrromethane [BODIPY(BDP)], PEGylated pH-sensitive
N,
N-dimethylacrylamide and thermosensitive N-isopropylacrylamide (PPDN), and rGO. Within the nanocomposite, rGO quenched the emission of [BODIPY(BDP)] fluorescence at a normal pH of 7.4, but not at a pH of 6, which mimicked a tumor microenvironment, or a pH of 5, which mimicked lysosomes. Accordingly, the complex displayed intense fluorescence in the lysosomes of human MDA-MB 231 breast cancer cells. Therefore, the complex has potential not only as an antitumor therapy mediated by DOX, but also as a fluorescent probe to identify cancer cells [
84].
In the aforementioned research by Tian et al., GO was used to quench the fluorescence of the photosensitizer Ce6 within a nanocomposite composed of Ce6-Pep, DSPE-PEG2000-FA, and GO. Upon entering the lysosomes of HeLa cells, cathepsin B cleaved the peptide to release the fluorescent Ce6 from GO. The lysosomal pH also stimulated the discharge of Ce6 from the nanocomposite. The nanocomposite was internalized by cancer cells in mice bearing HeLa tumors via folate receptor-mediated endocytosis and displayed strong fluorescence after entering their lysosomes, enabling the identification of cancer cells in vivo [
16].
Finally, in the previously mentioned study by Wu and colleagues, graphene from the smart vehicle GPCP quenched the fluorescence of the photosensitizer ICG. When ICG was released in lysosomes due to the acid-induced charge reversal effect, it began to fluoresce. Since the complex preferentially accumulated in tumor tissue, fluorescent tumors were readily observed in the MDA-MB-231 tumor-bearing mice [
12].
It is worth mentioning that some studies discussed earlier have used GNMs for imaging purposes. Fluorescence not only plays a crucial role in the detection of tumor cells but can also be important in determining the exact time and location of irradiation in PTT and PDT.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15071846