Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Hematology
Myeloid leukemia of Down syndrome (ML-DS) is characterized by a distinct natural history and is classified by the World Health Organization (WHO) as an independent entity, occurring with unique clinical and molecular features. The presence of a long preleukemic, myelodysplastic phase, called transient abnormal myelopoiesis (TAM), precedes the initiation of ML-DS and is defined by unusual chromosomal findings.
- Down syndrome
- myeloid leukemia
- acute megakaryoblastic leukemia
1. Introduction
Trisomy 21, the presence of a supernumerary chromosome 21, has been first described as the genetic cause of Down syndrome (DS) in 1959 [1]. Today, it is well established that DS constitutes one of the most frequent and genetically complex chromosomal disorders that are compatible with post-term survival [2]. In the majority of cases, constitutive trisomy 21 results from nondisjunction of Homo sapiens chromosome 21 (HSA21) during meiosis. The presence of all or a portion of a third copy of HSA21 is identified among the most common forms of mental retardation and is thought to be the cause of other medical conditions that affect multiple body systems [2]. The DS phenotype has been estimated to occur in approximately one in every 700 infants in the United States, with an overall prevalence of 10 per 10,000 births in Europe [3]. Due to raised awareness on the maternal-age-related higher risk of developing the syndrome, this prevalence is now decreasing [4]. Although life expectancy of individuals with DS has been extremely improved during recent decades (50–55 years), mortality among them remains approximately 5–10 times higher than mortality in the general population [3]. Patients with constitutional trisomy 21 present with a distinct collection of multiple clinical manifestations, as the extra genetic material results in the phenotypic expression of specific facial characteristics, as well as various malformations, congenital heart defects, immune and endocrine dysfunction, visual and hearing impairment, hematological abnormalities, and Alzheimer’s disease [4].
Almost all neonates with DS develop quantitative and morphological hematological disorders, yet still only 5–10% of them present with one of the preleukemic or leukemic conditions associated with DS [3]. Myeloid leukemia of Down syndrome (ML-DS) is identified as a distinct form of acute myeloid leukemia (AML), with a 46- to 83-fold increased incidence in DS children [5]. It is characterized by a long preleukemic stage, which is called transient abnormal myelopoiesis (TAM). It is marked by the presence of mutations on the GATA1 gene and is defined by its myeloproliferative nature affecting the megakaryocytic and erythroid lineages [6]. Both TAM and ML-DS present with the clinical and hematologic features of acute megakaryoblastic leukemia (AMKL) and have similar biologic behavior between them that is independent of the blast cell count [5]. AMKL is a rare subtype of AML and is classified as M7, according to the French–American–British (FAB) system. AMKL is defined as a form of leukemia with >20% blasts, of which 50% or more are of the megakaryocyte lineage, and it is associated with extensive myelofibrosis [7]. It is characterized by the presence of megakaryocytic antigens demonstrated by flow cytometry and immunohistochemistry [8]. In terms of blast morphology and immunophenotype, M7 blasts often resemble lymphoblasts, while one or more platelet glycoproteins are expressed on megakaryoblasts, namely CD41 (glycoprotein IIb/IIIa) and/or CD61 (glycoprotein IIIa) [8].
DS children have a 50-fold increased incidence of acute leukemia during the first 5 years of life. The acute leukemias in approximately 60% of affected DS children are myeloid, with at least 50% of these being AMKL [6]. The median age of patients with TAM is 1–1.8 years [4]. DS children with ML–DS have better prognosis compared with non-DS children with a myeloid neoplasm, with 5-year overall survival (OS) of 89–93% [6]. In most cases, TAM spontaneously resolves within 1 to 2 months after birth [5]. However, a percentage of 20–30% of TAM infants will develop ML-DS within the first 4 years of life [6]. It is important to identify and accurately diagnose TAM cases, which has proven challenging due to the absence of clear clinical, hematologic and molecular diagnostic criteria for this condition.
The World Health Organization (WHO) Classification of Tumors of Hematopoietic and Lymphoid Tissues defines TAM as the ‘increased peripheral blood blast cells in a neonate with Down syndrome’, without specifying the percentage blast count considered abnormal [9]. TAM diagnosis requires the presence of GATA1 mutations together with increased blasts and/or clinical features suggestive of TAM in a neonate with constitutional trisomy 21 [5,6,10]. Regarding TAM clinical presentation, this varies from multiple severe and life-threatening manifestations to asymptomatic disease where TAM may be diagnosed by various nonspecific blood count abnormalities. These include reduced platelet and increased leukocyte counts, as well as peripheral blood blasts [6]. Examination of a peripheral blood film is suggested to be made during the first week of life, as the percentage of circulating blasts often falls rapidly after this period [10]. TAM cells, from their origin in fetal liver, spread throughout the body, infiltrating the liver, pleural and pericardial spaces, skin and, in some cases, bone marrow [10]. This presents as hepatomegaly/hepatopathy (raised transaminases with conjugated hyperbilirubinaemia), as malignant effusions in pericardial and pleural spaces, and as a skin rash due to deposits containing TAM cells in the skin [10]. These and other clinical features—namely splenomegaly found in 30% of cases often because of portal venous obstruction, organomegaly, hepatic fibrosis, hyperleukocytosis, coagulopathy, and multi organ failure—are not entirely specific to TAM, because each of these features may also be found in non-TAM cases [10]. Thus, it is possible that asymptomatic TAM may not be diagnosed in some neonates, while in others, TAM may be overdiagnosed.
2. Genetic Landscape of Down Syndrome-Related Myeloid Leukemia
TAM is driven by mutations in the hematopoietic transcription factor gene GATA1, which result in a truncated isoform (GATA1s protein). These mutations are only seen in conjunction with trisomy 21, either constitutional or acquired [11]. This means that even in rare cases of myeloid leukemia in children without DS, the molecular pathogenesis and clinical outcomes are similar to those with DS [6]. These patients acquire trisomy 21, and such cases have already been well documented [12,13]. In addition, GATA1 mutations could be acquired or even germline [6]. All these findings constitute an interesting field of research, as it has been shown that the cooperation between these two lesions—trisomy 21 and GATA1 mutations—can lead to myeloid proliferation, and the clinical presentation of this condition can be independent of the order of their acquisition [6].
3. The Multistep Pathogenesis of ML-DS Development
The pathogenesis of ML-DS is considered to be the result of a multistep clonal evolution process. In the first place, individuals with trisomy 21 have a profound dosage imbalance in the hematopoiesis-governing genes located on chromosome 21 and thus are subject to impaired fetal as well as neonatal erythro-megakaryopoiesis [4]. When these individuals acquire GATA1 mutations, hematopoietic deregulation is observed, and a selective growth advantage to the leukemic cells is promoted [4]. Further transformation to ML-DS happens when additional mutations affecting chromatin and epigenetic regulators, as well as signaling mediators, contribute to further clonal evolution (Figure 1) [14].
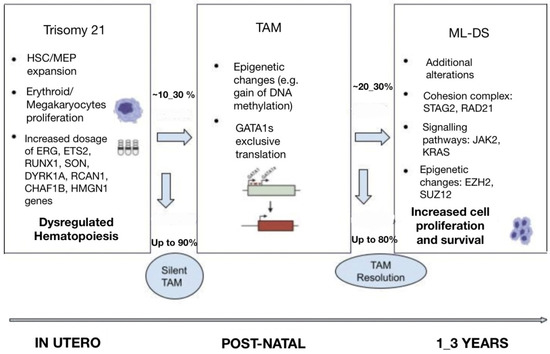
Figure 1. Multi-step pathogenesis of ML-DS.
3.1. Trisomy 21
It is well established that gains and losses of entire chromosomes or specific genomic regions are hallmarks of cancer. Neonates with gain of chromosome 21 are considered to be more frequently susceptible to hematological malignancies. Constitutive trisomy 21 results in altered hematopoiesis and dysregulated development of the megakaryocytic, erythroid and B-cell lineages [6]. Several studies from genetically engineered models have shown perturbed fetal hematopoiesis in DS individuals and have allowed for the identification of chromosome 21 dosage-sensitive genes [15]. It has been revealed that there is an increased proportion of hematopoietic stem cells (HSCs) and megakaryocyte–erythroid progenitors (MEP) in trisomy 21 fetal livers, which in cooperation with GATA1s expression, results in promoting blast and megakaryocyte expansion [15]. Moreover, studies have established fetal hematopoiesis from trisomic DS-derived induced pluripotent stem cells (iPSCs), which lead to significant expansion of myeloid and megakaryocytic progenitors compared with disomic cells [15]. More importantly, GATA1s expression correlates with defective embryonic hematopoiesis with a strong bias toward the myelo-megakaryocytic compartment and results in the development of TAM during fetal life [14].
The genes on chromosome 21 have been identified to play a predominant role in myeloid differentiation, and their encoded molecules are the following: transcription factors (ETS-related gene (ERG), ETS proto-oncogene 2 (ETS2), runt-related transcription factor 1 (RUNX1), SON DNA and RNA binding protein (SON)), signaling effectors (dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A), regulator of calcineurin 1 (RCAN1)), epigenetic modulators (chromatin assembly factor 1 subunit B (CHAF1B), high mobility group nucleosome binding domain 1 (HMGN1)), and a subset of miRNAs [14,15,16].
3.1.1. Transcription Factors
ERG and ETS2 are ETS-transcription factors and megakaryocytes oncogenes [6]. The involvement of ERG in leukemogenesis is facilitated by its overexpression, which causes megakaryoblastic expansion [6]. On the other hand, ETS2’s role is to promote and regulate megakaryopoiesis. Both ERG and ETS2 strongly cooperate with GATA1s protein to drive TAM and/or ML-DS development [14,15]. RUNX1 transcription factor is involved in the regulation of megakaryocytic differentiation. RUNX1 accelerates early hematopoiesis in the context of trisomy 21, and in correlation with ERG, ETS2 and GATA1s promote leukemogenesis [15,17]. SON is a transcription-factor-encoding gene located on HSA21, which plays a crucial role in proper blood cell formation, as it regulates the repression of megakaryocytic differentiation [18].
3.1.2. Signaling Effectors
The signaling molecule DYRK1A participates in multiple cellular functions through phosphorylation of the nuclear factor of activated T-cells (NFAT) and other substrates [6]. When the DYRK1A gene is overexpressed in murine models, the suppression of NFAT leads to increased megakaryoblastic proliferation [15]. In a corresponding manner, overexpression of RCAN1—an endogenous calcineurin inhibitor—represses the calcineurin-NFAT pathway and promotes excessive megakaryopoiesis [14,19].
3.1.3. Epigenetic Modulators
An epigenetic modulator coded by a gene on HSA21 is called CHAF1B. This is essential for normal hematopoiesis, and its overexpression impairs myeloid differentiation and promotes myeloid leukemia through binding of chromatin and interference with transcription factors such as CEBPA [20]. Another epigenetic modulator is HMGN1, which is the chromatin accessibility regulator. When upregulated, HMGN1 blocks myeloid differentiation and increases clonal progenitor expansion [21].
3.2. GATA1 Mutations
The GATA1 gene is encoded on the X chromosome. It is expressed in erythroid, megakaryocytic, eosinophilic, and mast cells, and it serves as a requirement for the proper growth and maturation of erythroid cells and megakaryocytes [4]. In the case that someone acquires GATA1 mutations, their megakaryocytes proliferate excessively and do not generate functional platelets [4]. Over one hundred GATA1 mutations have been reported in individuals with DS, which mainly constitute insertions, deletions, or duplications [15]. In the majority of cases, they occur in exon 2 of the GATA1 gene and lead to a short isoform of the GATA1 protein (GATA1s), which lacks the amino-terminal activation domain and subsequently creates an early stop codon [6]. Given that the GATA1 gene plays an essential role in the development of erythroid and megakaryocytic lineages, it is understandable that GATA1 mutations drive several functional and molecular changes on the regulation of erythro-megakaryocytic progenitors, namely progenitor expansion and disruption of erythroid and megakaryocytic differentiation [6,13]. In the context of trisomy 21, GATA1 mutations are strengthened and become sufficient to lead to TAM and/or ML-DS, whereas these mutations are not detected when TAM resolves [6]. The unique cooperation between GATA1 mutations and trisomy 21 in the evolution of TAM has been approved by the fact that GATA1 mutations have, to date, been discovered in nearly all patients with TAM and ML–DS. The lack of detected GATA1 mutations in ML-DS individuals is thought to be due to technical and sample limitations [15]. On the other hand, synergy between GATA1 mutations and subsequent additional chromosomal and genetic alterations is vital for the transformation of TAM to ML-DS, without knowing the exact features that would predominantly predict such transformation [13,14]. Some of the predictors used in clinical practice to decide the possibility of TAM progression to ML-DS include the detection of minimal residual disease (MRD) by flow cytometry (blasts > 0.1%), the persistence of GATA1 mutations beyond 12 weeks from the initial diagnosis, and the presence of thrombocytopenia (platelet count < 100 × 109/L) [6].
3.3. Additional Mutations
Additional somatic mutations are considered to affect genes encoding the cohesin complex, JAK kinases, and epigenetic regulators. Such additional mutations are rarely detected in TAM cases, in which patients carry only GATA1 mutations. It is suggested that ML-DS progression is mainly caused by activated signaling pathways in cooperation with deregulated epigenetic processes [6].
3.3.1. Cohesin Complex and Associated Components
The cohesin complex is a multi-subunit complex that surrounds chromosomal DNA and regulates its functions, namely sister chromatid cohesion, chromatin remodeling, transcriptional regulation, and DNA damage repair [6]. The mutations affecting the cohesin complex’s core subunits and its modulators are observed with a high occurrence in myeloid malignancies, and they cooperate with GATA1 mutations and the background of trisomy 21 to drive ML-DS [22]. The cohesin complex’s modulators STAG2 and RAD21 have been described to be highly prevalent in ML-DS [4,6]. Studies on DS-derived human iPSCs have shown that the consecutive introduction of GATA1 and STAG2 mutations in iPSCs lines result in the disruption of megakaryocyte differentiation and the expansion of the megakaryocytic population [22]. STAG2 deletion shows as a consequence a decrease in cell growth and proliferation and an increase in cell invasion and metastasis [6,23].
3.3.2. Signaling Pathways
It has been established that the majority of the mutations affecting and activating signaling pathways occur in genes encoding JAK family kinases, with the most commonly mutated genes being JAK2 and JAK3 [22]. Specifically, the expression of JAK3 genes is made in myeloid and lymphoid cells, and their mutations present with higher frequency in ML-DS individuals [6]. JAK kinases mediate downstream of thrombopoietin (TPO) and granulocyte–macrophage colony-stimulating factor (GM-CSF), and the JAK-STAT signaling pathway controls molecular and cellular processes, such as cell proliferation, differentiation, apoptosis, inflammation, and blood production [22]. In the presence of JAK3 mutations, normal TPO-mediated STAT5 activation is inhibited; thus, megakaryopoiesis is dysregulated and repressed [6,22]. The sequence of these events consecutively contributes to leukemogenesis.
Mutations in RAS oncogenes (KRAS, NRAS, NF1, PTPN11) have been associated with uncontrolled cell growth and colony formation, without sufficient clarity to how the mutated genes mediate leukemogenesis and cooperate with trisomy 21, GATA1 mutations and other additional mutations in the cohesin complex or the epigenetic regulators to drive ML-DS [4].
3.3.3. Epigenetic Regulators
Multiple epigenetic regulators have been described to contribute to leukemogenesis [6]. Among them, the most commonly presented are: additional sex combs-like 1 (ASXL1), BCL6 corepressor (BCOR), DNMT1, DNMT3A, embryonic ectoderm development (EED), E1A binding protein P300 (EP300), EZH2, KAT8 regulatory NSL complex subunit 1 (KANSL1), lysine demethylase 6A (KDM6A), lysine methyltransferase 2C (KMT2C), N-acetyltransferase 6 (NAT6), SUZ12, and tet methylcytosine dioxygenase 2 (TET2) [22,23]. EZH2 acts together with SUZ12, and they form the polycomb repressive complex 2 (PRC2). Both EZH2 and SUZ12 work as tumor suppressors and chromatin modifiers; hence, the mutated loss of their function and subsequent lack of PRC2 subunits’ function lead to blockage of megakaryocytic differentiation and acceleration of megakaryocytic proliferation [22,23].
Despite all these advances in understanding the underlying mechanisms associated with ML-DS presentation, there is still debate on the exact treatment plan for these patients, while little is yet known on the relapse of the disease, and the management of relapsed patients remains challenging [14].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15133265
This entry is offline, you can click here to edit this entry!