1. Introduction
Despite the significant progress in understanding the pathophysiology of Parkinson’s disease (PD), its prevention and treatment remain without significant therapeutic advancement. PD is a prevalent neurodegenerative illness resulting in progressive motor impairment and cognitive dysfunction [
1]. Most PD cases are sporadic, and only a low percentage is related to mutations in a few genes, causing familial PD [
2,
3,
4,
5]. As in other prevalent neurodegenerative disorders, aging is the principal risk factor for developing this condition [
6]. Toxic compounds and genetic mutations likely play a central function in PD development and progression [
2,
3,
4]. However, the pathophysiology of PD onset and progression is not fully understood.
PD patients develop a progressive loss of dopaminergic neurons in the substantia nigra of the brain, exhibiting neuronal Lewy bodies formed by protein aggregates whose principal component is α-synuclein [
7,
8]. The α-synuclein accumulation seems to start as microaggregates at the presynaptic level, likely interfering with neurotransmitter vesicle trafficking and release in dopaminergic cells [
9,
10]. However, Lewy bodies also appear in asymptomatic aged people [
11], indicating they are assembled in aggresome-related processes to rescue neurons from protein misfolding [
12].
The molecular intricacy of PD denotes that multiple biochemical pathways underlie the clinical manifestations and progression of the disease [
2,
3,
4,
5], expressing the involvement of oxidative damage, reduced antioxidant ability, and mitochondrial dysfunctions related to cellular senescence in dopaminergic neurons [
13,
14,
15].
2. Role of Glutathione Precursors in Parkinson’s Disease
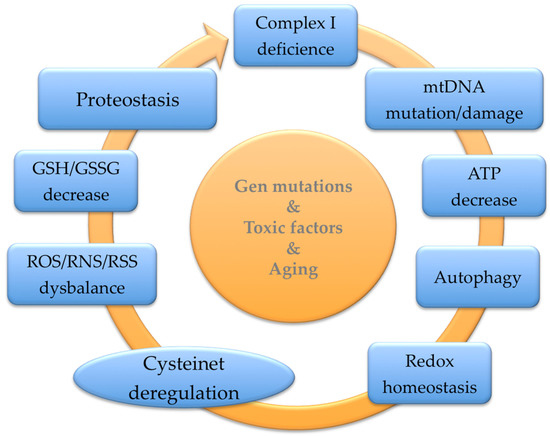
Redox modifications of sensitive cysteines sustain the thiol quality control of essential proteins, supporting cells against oxidative and xenobiotics damage (
Figure 1). Cysteine is the rate-limiting substrate for the cellular tripeptide GSH (cysteine, glycine, and glutamic acid) synthesis. Cystine arrives in the cell via the xCT
(−) CSSG/L-glutamate antiporter (SLC7A11) system for maintaining GSH synthesis [
33]. Despite low levels of H
2S compared with other low-molecular-weight thiols, H
2S is a powerful thiol working via the thioredoxin or NADPH systems to play its reducing effects [
161]. Cells exposed to elevated H
2S present higher GSH levels than the nontreated control, indicating an enhanced GSH recycling rate [
162]. These data show that H
2S maintains the cellular redox equilibrium defending cells from oxidative damage in GSH deficiency conditions [
162].
Figure 1. Summary of the main pathophysiological mechanisms that may be improved with GSH precursors.
GSH participates in the reduction of oxidative stress, the maintenance of redox equilibrium, the improvement of metabolic and organic pollutant detoxification, and the regulation of the immune system [
17] (
Figure 1). Clinical research indicates that nutritional interventions, including phytochemicals and foods, have pivotal effects on GSH’s proper balance. Moreover, individual genetic variability modifies the GSH levels influencing the global GSH status due to variability in enzymes implicated in its synthesis and renewal. Red blood cell GSH has a broad intra-individual divergence, though it is relatively long-lasting because of the variation the in genes controlling GSH concentrations [
163].
2.1. Nutrients and GSH Status
2.1.1. Amino Acids, Peptides, and Proteins
Many nutrients contribute to sustaining optimal GSH status. The intake of proteins affects the amino acid pool for synthesizing GSH. Indeed, supplementation with proteins with high cysteine content, such as whey protein, results in a dose-dependent boost in lymphocyte GSH concentrations [
164,
165]. Serine may positively impact GSH generation, increasing cysteine availability and reducing hyper-methylation, supporting the glycine metabolism for GSH synthesis.
GSH is easily oxidized, showing a short half-life in plasma (<3 min). Moreover, GSH does not efficiently cross cell membranes requiring elevated quantities to gain therapeutic concentrations [
166]. Although digestive peptidases can degrade oral GSH showing no change in GSH concentrations or oxidative stress parameters [
167], some clinical studies found substantial increases in the body’s GSH stores under different administrations. For example, using GSH monoesters improved its bioavailability [
168].
2.1.2. Vitamins
Some vitamins participate in the GSH status. Low doses of vitamin C can increase GSH levels in red blood cells [
169]. Vitamin E administration may also increase GSH levels by reducing oxidative stress markers [
170,
171]. B2 vitamin is a necessary coenzyme for glutathione reductase, which restores the GSH from GSSG [
172]. Moreover, the B5 vitamin may support GSH status via ATP generation [
172]. Finally, alpha-lipoic acid can restore antioxidant capacity, including GSH regeneration [
173,
174].
2.1.3. Flavonoids and Thiol-Rich Compounds
Many phytonutrients participate in the cellular antioxidant defenses that boost GSH metabolism, including foods containing all three thiol-rich compounds (GSH, NAC, and cysteine) [
175]. NAC is concentrated in plants of the Allium species, especially in the onion. Therefore, eating a diet rich in asparagus, onion, cucumber, grapefruit, green and red pepper, strawberry, and tomato may easily contribute to redox homeostasis [
175]. Xanthohumol is a flavonoid that contains an isoprenyl group that can regulate the Nrf2-ARE pathway in neuronal cells by covalently modifying cysteine residues in proteins. It can change the activity of glucose-6-phosphate dehydrogenase (G6PD), limiting the production of NADPH [
176]. Xanthohumol can also hamper the generation of inflammatory intermediates like NO, IL-1β, and TNF-α, interfering with the activation of the NF-κB pathway [
177].
2.1.4. Michael Acceptor Molecules (MAMs)
In physiological circumstances, thiols and amines in small molecules or proteins can undergo a Michael addition reaction [
178]. Diverse MAMs derived from plants have active electrophilic groups that target reactive residues on proteins with functional biological effects and low toxicity, providing some therapeutic options. Cysteine, homocysteine, GSH, and SCCPs are widespread reactive thiols that can interact with MAMs. MAMs can activate the Keap1-Nrf2-ARE pathway, playing antioxidant functions by covalently binding to sensitive cysteines. They also have anti-inflammatory functions through the inhibition of the NF-κB, a crucial sensitive cysteine protein modulated through redox reactions. These substances, used as dietary supplements, are effective for diverse oxidative stress-mediated inflammatory and developmental and degenerative diseases [
179].
MAM-derived molecules such as andrographolide and lophirone B and C can modify the Keap1–Nrf2 pathway by alkylating specific reactive cysteines in Keap1, enhancing the cellular antioxidant ability and liver detoxification enzyme activities [
180,
181]. Moreover, they may enable the expression of NQO1, HO-1, UGT, SOD, GST, and EPH, improving cellular antioxidant and liver detoxification capacities [
180]. Finally, Cardamonin can alter the functional cysteines of Keap1 through α, β-unsaturated carbonyl structure forming a covalent adduct, promoting Nrf2 nuclear translocation and triggering the expression of downstream genes of ARE [
181]. Interestingly, Cardamonin can enter the blood–brain barrier, exerting a potential neuroprotective influence on ROS-associated neurodegenerative disorders ([
181].
The α,β-unsaturated arrangements of MAMs can hamper the inhibitor of nuclear factor-kappa B kinase (IKK) by attaching to the reactive cysteines placed in the activation loop of IKKβ, inhibiting IκBα phosphorylation and therefore blocking the NF-κB route [
180]. Moreover, physalin A can target Cys59, Cys179, Cys299, Cys370, Cys412, and Cys618 residues in the IKKβ loop resulting in anti-inflammatory effects [
182]. Additionally, PKC has a cysteine-rich region and is the upstream enhancer of MAPK, the principal IKK activation signal [
183]. Also, the α,β-unsaturated structure may interfere with the NF-κB pathway by linking to PKC cysteines [
178]. Finally, Helenalin can interfere with the NF-κB route by binding to Cys38 of p65 [
184].
2.2. CoQ10-Related Compounds
EPI-743 is a synthetic fat-soluble drug (para-benzoquinone) that is much more potent than CoQ10 or idebenone in protecting cells against oxidative damage in mitochondrial diseases [
185]. Indeed, EPI-743 represents a new treatment for inherited mitochondrial respiratory chain disorders [
186,
187], and it is an antioxidant that crosses the blood–brain barrier, increasing glutathione synthesis [
186,
188]. Also, EPI-743 is used for some fatal inherited neurodegenerative disorders [
189]. Compared to other antioxidant molecules, EPI-743 probably exerts a more intricate action, modulating multiple cell pathways, including Nrf2, a crucial cellular regulator against oxidative damage. Moreover, EPI-743 has shown improvement against oxidative stress markers that correlated with clinical improvement and a significant decrease in CNS glutamine/glutamate levels in PD patients [
190]. These findings were associated with improvements in UPDRS scores that approached statistical significance [
190].
2.3. N-Acetyl-Cysteine (NAC)
2.3.1. NAC Bioavailability and Safety
Maintaining redox regulation at the protein level is challenging due to the numerous metabolic and cell signaling pathways involved [
17,
20,
21,
22,
23,
24,
41]. Any compound that can interfere with the redox proteome must be considered a powerful medication, even at low doses. Unlike GSH, NAC has better oral and topical bioavailability, and has been commercially accessible for a long time [
17]. The research suggested that NAC regulates SCCPs in a comprehensive range of pathways involved in neurodegenerative and psychiatric disorders [
17,
20,
21,
22,
23,
24,
41,
191,
192,
193,
194,
195,
196,
197,
198,
199,
200,
201,
202,
203]. Moreover, NAC may work synergistically with other supplemental nutrients, such as glycine [
204]. Although there is still some controversy about the doses, NAC is a supplement that increases GSH levels contributing to decreasing oxidative damage. Since NAC is a membrane-permeable cysteine precursor, it does not need the alanine–serine–cysteine system to enter the cell, yielding cysteine to regenerate total GSH content and reducing excessive GSSG (
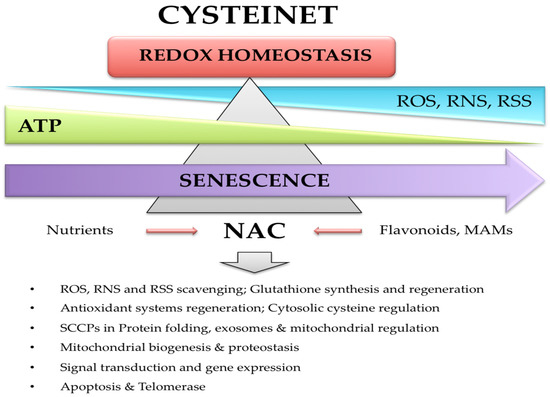
Figure 2).
Figure 2. Cysteinet deregulation in aging and Parkinson’s disease. Senescence is associated with a deregulation of reactive oxygen (ROS), nitrogen (RNS), and sulfide (RSS) species. Moreover, the mitochondrial bioenergetic ability is impaired, contributing to glutathione (GSH) decline and cysteinet deregulation. Chronic NAC supplementation may rejuvenate cysteinet and redox homeostasis in Parkinson’s disease.
A unique intravenous dose of NAC augmented the blood GSH/GSSG ratio and total GSH in the brain, showing a positive direct correlation between the blood ratio and brain GSH concentrations [
205]. However, oral NAC intake is quickly absorbed with a plasma half-life of 2.5 h and no detectable NAC in 10–12 h. Single doses of 600 mg/day and 1200 mg/day result in 16 mM and 35 mM plasma concentrations, respectively [
206]. The terminal half-life of reduced NAC is 6.25 h, incorporated into proteins after several hours [
206]. There is sufficient clinical evidence of the beneficial action of NAC against oxidative damage in multiple conditions, including oncological and cardiovascular diseases, ophthalmic illnesses, HIV infection, metal toxicity, cerebral ischemic and bleeding disturbances, traumatic brain injury, and neuropsychiatric disorders [
207,
208,
209,
210,
211,
212]. Low NAC supplementation may prevent brain aging and neurodegenerative illnesses, not only via classical antioxidant mechanisms but rejuvenating the cellular redox homeostasis via SCCPs regulation [
17,
20,
21,
22,
23,
24,
41].
NAC has shown beneficial effects in neurodegenerative diseases through GSH replenishment and its direct scavenging ability against reactive species [
17]. However, its potential significance in modulating the redox proteome and proteostasis is vaguely known. NAC can rejuvenate the specific activities of complexes I, IV, and V in mice presynaptic mitochondria from aged mice. These effects are attributed to the repair of functional cysteines [
213,
214,
215,
216]. Moreover, in vivo enzymatic complexes’ specific activities were corrected by chronic NAC supplementation, improving ATP and GSH levels and reducing lipid and protein oxidation markers in pre-synapses [
213,
214,
215,
216].
2.3.2. NAC in Cysteinet Regulation
NAC may modulate the redox homeostasis of functional cysteines in numerous proteins, restoring critical cellular pathways to support neuronal survival [
24] (
Figure 2). For example, NAC activates the Ras-ERK pathway in PC12 cells, preventing neuronal death that lacks trophic factors through nonantioxidant mechanisms. Ras proteins have essential redox-sensitive cysteines suggesting that NAC directly activates Ras via its reducing ability [
217,
218]. NAC can also protect human neurons against apoptosis induced by Aβamyloid 1–42 [
193], starting the p35/Cdk5 pathway and decreasing the phosphorylation/deactivation of MLK3–MKK7–JNK3 signaling (reviewed in [
17]). NAC can suppress transcription factors such as NF-κB, inhibiting the next cytokine generation [
219]. Moreover, NAC may downregulate the APP gene transcription in neuroblastoma cells by lowering the activity of NF-κB [
220]. These NAC effects can be due, at least partially, to sensitive cysteines in those SCCPs [
17,
24] (
Figure 1 and
Figure 2).
Redox metabolism regulates transcription factors like NF-κB, AP-1, and the IKK, which include redox-sensitive cysteines [
221]. Specifically, NF-κB has Cys38 and Cys62 essential for its function [
134,
221], IKK possesses Cys179 that intervenes in the kinase activity [
222], and the transcription factor AP-1 binds DNA through functional cysteines [
223]. Interestingly, NAC can control transcription [
214]. NAC can decrease the oxidative damage of NF-κB in clinical sepsis and other oxidative stressing conditions [
219,
224].
NAC can cross the blood–brain barrier interfering with the main SCCPs in the brain, balancing cysteinet deregulation by replenishing cellular-soluble (H
2S, cysteine/glutathione) and protein-associated thiols, restoring the mitochondrial bioenergetic power and biogenesis. In addition, NAC increases the level of brain-derived neurotrophic factors sustaining the survival of neurons and stimulating neurogenesis [
225,
226]. Therefore, NAC must be considered a potent compound, even at low doses, modulating essential protein function from multiple pathways, and consequently, its supplementation has to be accurately specified.
2.3.3. NAC in Protein Misfolding
NAC may rejuvenate age-related protein misfolding and aggregation by stopping cysteines’ oxidative damage. Proteins can suffer conformational modifications when they suffer oxidative injury [
227]. In senescence, the balance among protein synthesis, folding, and clearance may shift towards misfolding and aggregation, contributing to neurodegeneration. Cysteine inhibits the assembly and accumulation of amyloidogenic peptides [
228], and the beneficial effect of NAC on protein aggregation was confirmed in a mouse model of Huntington’s disease [
229]. The changes in the 3-D structure of proteins occur when they accumulate in tissues, mainly in proteins with repetitive amino acids, like polyglutamine in Huntington’s disease [
229]. Therefore, NAC can partially fix the aggregation of protein neutralizing misfolding mechanisms in neurodegenerative disorders. Accordingly, cysteine inhibits the fibrillation of Aβ1–40 and Aβ1–42, and their cysteine-induced aggregates were less toxic than those generated by catechin [
228].
5.3.4. NAC in Cellular Vesicle Regulation and Signaling
The oxidative control of reactive cysteines on proteins can drive exosomes’ formation and functions. NAC supplementation can rejuvenate vesicular secretion and composition [
57]. Thus, a beneficial mechanism of NAC administration in PD may be the replenishment of cysteine thiols to restore the redox regulation of extracellular vesicles. NAC and N-acetylcysteine amide (NACA) can suppress exosome building by scavenging thiol groups, preventing their reaction with cellular thiols. Specifically, NAC may repair exosome secretion, composition, and functions under stressing conditions to physiological levels, controlling oxidative modifications in exosome signaling and aging [
230,
231].
On the other hand, CSPα is an SCCP that participates in synaptic vesicle endocytosis and synaptic neurotransmission [
232]. CSPα oligomerization relies on a cysteine-rich domain used for attachment to synaptic vesicles [
232]. The α-synuclein also functions as a chaperone-like protein in combination with CSPα for building the SNARE complex [
233]. This α-synuclein process concerns the association with synaptotagmin, which have reactive cysteines in their structure [
233,
234].
Therefore, thiol-supplying substances may protect against the detrimental disturbance of cellular vesicle formation under oxidative states, supporting the suitability of NAC as a preventive and restorative compound against neurodegeneration in aging and associated disorders via cysteinet rejuvenation.
2.3.5. NAC and Mitochondria
Oxidative damage in synaptic mitochondria is associated with brain senescence [
213,
214,
215,
216], likely affecting critical cysteines in mitochondrial proteins. The research think the redox equalization of mitochondrial SCCPs may explain many effects of NAC chronic implementation at low doses in the brain [
17,
24]. Investigations in presynaptic mitochondria from aged mice chronically supplemented with NAC indicated its rejuvenation properties, boosting ATP by activating mitochondrial respiration and oxidative phosphorylation, raising GSH levels, and diminishing lipid and protein oxidations [
20,
21,
22,
23,
24,
41,
193]. Adequate NAC levels in the aged brain maintain glutamate uptake via astrocytes and neurons expressing the excitatory amino acid carrier-1. NAC switched the GSH deficit and oxidative damage in the defective mouse of these carriers, indicating that the excitatory amino acid carrier-1 is required for cysteine uptake and GSH synthesis in the brain [
235].
2.3.6. NAC and Protein Kinases
NAC may rejuvenate vital pathways against neural death, such as the Ras–ERK (extracellular signal-regulated kinase) pathway, with nonantioxidant effects when trophic factors are absent [
17,
18,
24]. Ras possesses functional cysteines involved in redox-regulated mechanisms [
212,
213]. In this regard, NAC protects human cortical cerebral neurons against Aβ-amyloid 1-42 [
193], inducing p35/Cdk5 action and declining phosphorylation of the MLK3-MKK7-JNK3 signaling pathway. Cdk5 is a cyclin-dependent kinase [
236,
237,
238] involved in neuroprotection, neuronal migration, axonal guidance, and synaptic spine density [
239]. Cdk5 dysfunction participates in the pathophysiology of various neurodegenerative conditions, including PD [
240]. Cdk5 activation via proteolysis of p35 to p25 via calpain contributes to neuronal toxicity. Indeed, Cys83 and Cys157 s-nitrosylation triggers SNO-Cdk5 building, inducing β-amyloid dendritic spine drop [
240]. Also, SNO-Cdk5 is present in AD brains compared to controls, indicating that SNO-Cdk5 may disrupt its activity and thus participating in neuronal death [
240].
Mixed lineage kinase 3 (MLK3) is activated via s-nitrosylation at the sensitive Cys688, contributing to its activation and brain ischemia/reperfusion damage [
241,
242]. In this regard, NAC can inhibit MLK3 activation in early ischemia/reperfusion phases following brain hypoxia [
242]. Interestingly, MLK3 can phosphorylate other SCCPs, such as Pin1 [
243], increasing their function and translocation to the nucleus [
244].
2.3.7. NAC and Telomerase
Increased ROS may affect hTERT localization and activity, which are reverted by low doses of NAC, preventing hTERT translocation from the nucleus to the cytosol and endothelial cell aging [
245]. Interestingly, hTERT functionality can suffer post-transcriptional regulation via protein kinases such as c-Abl, PKC, ERK1/2, and Akt [
246], some of which are SCCPs. Therefore, they are susceptible to being modified by NAC. Another study indicates that chronic NAC therapy decreases cultured endothelial cell aging by hampering telomere erosion via partial hTERT activation, suggesting that the beneficial effect of NAC depends on the gain of the hTERT function [
247]. Also, NAC started the translocation and activity of the cytoplasm to the nucleus hTERT when aging was delayed [
247].
2.3.8. NAC-GSH and Nanotechnology
The supplementation of GSH and its precursors, such as cysteine, has some issues because of their instability and toxicity. In this regard, the usefulness of NAC lipophilic products enhanced their tissue biodistribution. Another alternative for improving GSH bioavailability is employing delivery systems such as liposomes, microemulsions, nanoparticles, and microparticles prepared with natural or synthetic polymers, with promising results in humans [
248]. There are also investigations using GSH to implement medications into the brain or to control medicine delivery in the intracellular compartment [
248].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12071373