Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
The role of triglyceride-rich lipoproteins (TRLs) and their remnants in atherosclerosis has come to the forefront in the past decade. Triglycerides (TGs) stand as markers of the remnants of the catabolism of TRLs that tend to contain twice as much cholesterol as compared to LDL. The accumulation of circulating TRLs and their partially lipolyzed derivatives, known as “remnants”, is caused mainly by ineffective triglyceride catabolism. These cholesterol-enriched remnant particles are hypothesized to contribute to atherogenesis.
- apoCIII
- chylomicrons
- VLDL
- LDL
- apoB48
1. Introduction
The cornerstone for lowering the risk of atherosclerotic cardiovascular disease (ASCVD), for the past three decades, has been statin medication [1,2,3,4,5]. Numerous investigations conducted during this time have conclusively shown a link between low-density lipoprotein cholesterol (LDL-C) and ASCVD [1,3,6,7]. As a result, medications that reduce total lifetime exposure to LDL-C have been effective in reducing the risk of ASCVD. However, despite the effectiveness and affordability of statin medication, ASCVD continues to be the world’s leading cause of death. It is significant that many people using statin therapy still have a residual risk of developing ASCVD and cannot meet their target LDL-C targets [4,8,9,10,11,12,13,14,15,16,17]. This residual risk, which may amount to 50% after optimal statin treatment, is the consequence of both immunological and lipid disturbances [18,19,20,21,22,23]. Regarding the lipid disorders, the role of triglyceride-rich lipoproteins (TRLs) and their remnants has come to the forefront in the past decade [24,25,26,27]. Triglycerides (TGs) stand as markers of the lipoproteins that contain them, and more specifically of the remnants of the catabolism of TRLs, which tend to contain twice as much cholesterol as compared to LDL. The accumulation of circulating TRLs and their partially lipolyzed derivatives, known as “remnants”, is caused by ineffective intravascular TG metabolism and recapture. These cholesterol-enriched remnant particles are hypothesized to contribute to atherogenesis in addition to LDL [8,24,27,28,29,30,31,32,33,34,35].
Even though epidemiological studies have long associated plasma TG levels with the risk of atherosclerotic cardiovascular disease, the association between TG levels and other risk factors has led many researchers, but not all, to hypothesize that the relationship is confounded and likely not causal. Certainly, TGs per se are neutral from the standpoint of pure chemistry and are also neutral from the viewpoint of a direct pathological effect on atherogenesis.
This opinion, however, completely changed when it was discovered that variations in TG levels caused by genetics, but not by low high-density lipoprotein cholesterol (HDL-C) levels, were linked to ASCVD [24,25,27,28,36]. Additionally, clinical outcome studies conducted to evaluate the advantages of raising HDL-C levels failed to detect a decrease in the risk of cardiovascular events. Therefore, management of increased TRLs has emerged as the next prospective lipid-lowering method to minimize ASCVD risk, although low HDL-C levels are now regarded as an indicator of ASCVD risk (mainly because they are a marker of TRL decreased turnover) but not a target for therapy [3].
Chylomicron and very low-density lipoprotein (VLDL) intravascular catabolism results in a variety of remnant particles that have undergone partial lipolysis. Lipases, lipid transfer proteins, and the number of exchangeable lipoproteins all have an impact on the amounts and characteristics of these molecules in plasma [27,37,38,39,40,41,42,43,44,45,46,47]. Remnants may develop pathologic characteristics that aid in the progression of ASCVD, such as elevated cholesterol levels and the transport of thrombogenic and inflammatory mediators, during their plasma transit [7].
In the past decade, a plethora of information has emerged on the role of TRL metabolism and their resulting remnant particles in atherogenesis, which has also sparked the development of new therapeutical targets.
2. Origins of Circulating Triglyceride-Rich Lipoproteins
TGs provide an efficient and compact energy source. The development of metabolic processes to store and retain available TGs for usage in a controlled manner was required because humans have had relatively limited and sporadic access to food containing high quantities of fat for a large portion of their evolutionary history as hunter-gatherers. To effectively utilize this resource, highly regulated and efficient procedures are used to move TGs between sites of absorption (intestine), storage (adipose tissue), repackaging (liver), and usage (muscle). Circulating TRLs stem from the liver in the form of VLDLs carrying TGs produced in the body in an endogenous TRL pathway. TRLs coming from food are synthesized by the intestines in the form of chylomicrons [8,12,18,24,26,29,41].
2.1. Endogenous Pathways; VLDL Synthesis by the Liver Occurs throughout the Day and Is Critical during Fasting (Figure 1)
Triglycerides produced in the liver are transported by VLDL to the body’s peripheral tissues for utilization. These lipoproteins transport over 90% of the TGs in the bloodstream while fasting. Overall, the amount of fat transported in this way is over 50 g per day. These TGs come from fatty acids (FAs) from a variety of sources including chylomicron remnants, hepatic de novo lipogenesis (DNL, the process by which carbohydrates are transformed into FAs), uptake of non-esterified (free) fatty acids (FFAs) from the plasma, and release of FAs from hepatocyte cytosolic lipid droplets [1,24,30,40,41].
The process for assembling VLDL is intricate and strictly controlled. Researchers provide a simplified diagram (Figure 1A). Microsomal triglyceride transfer protein (MTTP) mediates the co-translational lipidation of the developing apoB polypeptide in the endoplasmic reticulum (ER), which is the first step in this route and forms a pre-VLDL particle. Pre-VLDL can be further lipidated to generate VLDL2, a lipoprotein with a tiny, triglyceride-rich core when the chaperones separate and the lipidation is sufficient to enable the proper folding of apoB (Figure 1A). ApoB100 is one of the largest known proteins, with a mass of 500 kDa. In normal conditions, but much increased in livers with excess fat (Figure 1B) large triglyceride-rich VLDL1 is created by fusing nascent VLDL2 with lipid droplets in the Golgi apparatus. This VLDL1 is then released into the bloodstream as well. About 45,000 triglyceride molecules per particle make up VLDL1, while just 10,000 TG molecules per particle make up VLDL2 [24]. Their metabolism is slightly different.
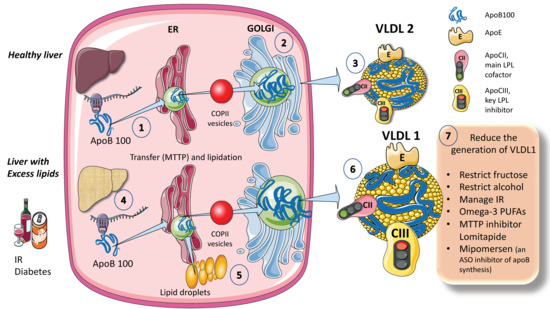
Figure 1. Production of endogenous triglyceride-rich lipoproteins (TRLs) by the liver. A. Healthy liver (1) ApoB100, one of the largest proteins, is synthesized by hepatocytes in the reticulum (ER) where it is moved along by the concourse of microsomal triglyceride transfer protein (MTTP). (2) Lipidated apo B is moved to the Golgi apparatus by means of COP II (Coat Protein Complex II) vesicles. (3) Finally, very low-density lipoprotein 2 (VLDL2), containing phospholipids and cholesterol besides TG, is secreted into the circulation. B. Liver with excess lipids. (4) TG employed for the synthesis of VLDL may come from re-esterified fatty acids catabolism of remnants or de novo lipogenesis (DNL). DNL implies the synthesis of fatty acids from glycerol and acetyl CoA derived from the metabolism of glucose and fructose. DNL is only 5% (of the total FA synthesis) in normal livers, but in (5) and (6) it reaches 25% when there is insulin resistance, diabetes, and high consumption of sugars or alcohol. Excess fat from DNL, build-up of circulating fatty acids, or liver steatosis is employed to enrich VLDL2 to a much larger particle called VLDL1. As compared to VLDL2, VLDL1 is not larger, but it contains more apoCIII. apoCIII is a very potent inhibitor of the catabolism of VLDL, thus promoting hypertriglyceridemia. The prolonged residence time favored by a delayed catabolism promotes the action of hepatic (HL) and endothelial lipase (EL) that ultimately results in the production of small and dense LDL, which is highly atherogenic. (7) Researchers summarize some of the dietary and pharmacological molecules that reduce the production of VLDL1. Unfortunately, but not surprisingly, the pharmacological agents result in the accumulation of TG in the liver, thus producing a fatty liver. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
When liver TG levels are high, the production of VLDL1 is accelerated, probably to shield hepatocytes from the harmful effects of TG overload (Figure 1B). VLDL1 synthesis is significantly influenced by insulin, a key regulator of metabolic processes related to energy storage and consumption (particularly, fat and carbohydrate metabolism). Insulin inhibits the release of VLDL1 from the liver both directly and indirectly (by reducing FFA flow into the liver). Direct inhibition of VLDL1 production occurs postprandially (when insulin levels are elevated). In people with insulin resistance (IR), elevated amounts of liver fat, and type 2 diabetes mellitus, both the direct and indirect effects of hormones are impaired [24,25,26,27,29]. Drinking alcohol causes a dose-dependent increase in hepatic FA production and a dose-dependent decrease in FA oxidation, which has the net effect of increasing VLDL secretion. The synthesis of VLDL2 is more tightly related to cholesterol metabolism and is not directly influenced by insulin. Endogenous cholesterol production is boosted when VLDL2 secretion is increased possibly to control hepatic cholesterol levels. VLDL2 rather than VLDL1 is generated in greater quantities in people with elevated plasma cholesterol levels, which may be a factor in the LDL-C overproduction that is frequently observed in these patients [8,27,34,35,42].
As shown in Figure 1, VLDL1 and VLDL2 contain several surface apolipoproteins other than apoB100, many of which are acquired in plasma, transferred from HDL. For the sake of researchers discussion the most important ones, are apoE, which is critical for the binding of these particles to receptors for uptake, apoCII, the main activator of lipoprotein lipase, and apoCIII, the main inhibitor. One main difference between VLDL1 and 2 is that VLDL1 contains more apoCIII [6,16,30].
2.2. Exogenous Pathways; Chylomicron Synthesis by Enterocytes Occurs in the Postprandial Period
In Figure 2, in a simplified outline, the main features of chylomicron production by enterocytes. The chylomicron assembly pathway is substantially less well understood than the liver VLDL assembly pathways, and it transports all fat in the diet (over 70 g per day) with the exception of short-chain fatty acids. Several theories have been put forth to explain how these enormous TRLs form. As shown in the figure, a fat load in food is digested by pancreatic lipase with the activation provided by liver co-lipase in a complex system of micelles that ultimately leads to the production of free fatty acids and monoglycerides [39,40]. After absorption by the enterocyte, these fatty acids are re-esterified into TG, which represents the first point of regulation of the composition of these molecules, i.e., the relatively amount of saturated versus mono and poly and saturated fatty acids. The backbone of chylomicrons is apoB48, which represents a truncated, spliced form of apoB100 that is missing from the LDL receptor binding site. In a similar, but less well-described process, apoB48 is lipidated with TG, phospholipids, and cholesterol through its passage across the ER and Golgi apparatus [48,49,50,51,52]. Large triglyceride-rich chylomicrons and primordial, lipid-poor, apoB48-containing lipoproteins may assemble through separate mechanisms. As shown in Figure 2, chylomicrons that have just been secreted are discharged into lymphatic channels and go to the thoracic duct, where they are transported with their load of dietary fats and fat-soluble vitamins to the left subclavian vein. Therefore, most dietary lipids do not enter the hepatic portal system like other nutrients do [50,52].
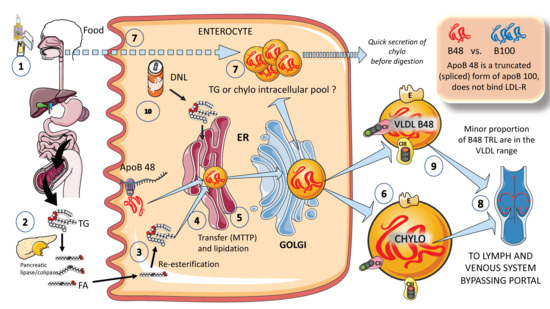
Figure 2. Production of exogenous triglyceride-rich lipoproteins (TRLs) by the liver: Chylomicrons and VLDL apoB48. (1) Lipids in the diet are (2) digested by pancreatic lipase, which is activated by liver co-lipase, in a complex system of micelles, resulting in the absorption of fatty acids and monoglycerides. (3) Fatty acids are re-esterified by the enterocyte and the resulting TGs (4) serve to lipidate apoB48. The latter is a truncated, spliced form of apoB100, which lacks the LDL receptor binding sites. (5) Much less is known about the transit and steps in chylomicron production as compared to VLDL. The resultant chylomicrons, which are very large particles, are (6) secreted into the circulation (7). However, very recently it has been shown that some of them are either stored as an intracellular pool or swiftly produced from TG droplets that can be called upon very quickly when the next meal comes, following signals stemming from the intake of more food and even before it reaches the intestines. (8) Chylomicrons secreted into the circulation contain phospholipids, cholesterol, and the liposoluble vitamins and are first transported by the lymph, reaching the venous, and finally the arterial circulation in a third step. (9) It has recently been shown that some of the apoB48-containing lipoproteins coming from the intestines are also in the VLDL size range. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Of note, contrary to what was previously believed, recent research has demonstrated that some TGs are retained and stored in the intestines, and that the first source of TGs produced after a meal may come from previous food intake [7,24,25]. This theory suggests that intestinal cells can start secreting stored TGs in chylomicrons immediately after meal consumption without the need to wait for dietary lipids to pass the enterocyte boundary. The taste-gut-brain axis has been connected to the release of chylomicrons, which may help to explain why fat or glucose need to be just tasted and not necessarily consumed to produce chylomicrons.
Finally, apoB 48-containing TRL is now known to be secreted both continuously at a low rate in the fasting state and at a greater rate postprandially. They are secreted not only as chylomicrons but also as smaller lipoproteins detected in the VLDL density range, Figure 2 [24,29].
A parallelism exists between the liver and the intestine as to which factors affect the production of chylomicrons. Indeed, FFAs and insulin both control chylomicron synthesis and release. In people with insulin sensitivity, insulin inhibits the release of apoB48-containing lipoproteins from the intestine, but in insulin-resistant conditions, such as metabolic syndrome (MetS) or type 2 diabetes, this regulatory function is compromised, leading to an overproduction of chylomicrons and apoB48-VLDL. Additionally, it appears that incretins play a significant role in altering chylomicron secretion via receptor-mediated pathways and neuronal networks [24,26,29,39].
This entry is adapted from the peer-reviewed paper 10.3390/jcm12134399
This entry is offline, you can click here to edit this entry!