Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cervical cancer (CC) is a major health problem in women of childbearing age. The immune response plays a crucial role in detecting and preventing the development of CC. However, factors such as age, prior or repeated human papillomavirus (HPV) infection, changes in the microbiota of the reproductive tract, and lifestyle choices can lead to immune dysregulation and increase the risk of CC. One of the critical components of the TIME is tumor-infiltrating lymphocytes (TILs), which are altered in CC and can contribute to tumor growth.
- cervix
- cervical cancer
- HPV
- immune cells
- inflammation
- TIME
1. Tumor-Associated Macrophages (TAMs)
Tumor-associated macrophages (TAMs) are another crucial component of the tumor immune microenvironment (TIME), and their levels can influence cancer prognosis depending on their diversity as determined through single-cell omics [1][2]. For example, they help in neoangiogenesis which helps in tumor growth, survival, and metastasis by releasing angiogenic factors, including vascular endothelial growth factor (VEGF) and creating a tumor-promoting immunosuppressive environment (Figure 1). There are two subgroups of macrophages, M1 and M2, with different roles in cancer progression [1][3]. M1 macrophages may be less detrimental to cancer prognosis, as they are more associated with phagocytosis and antitumor inflammation reactions, while M2 macrophages exhibit immunosuppressive, tumor-promoting activities by secreting different molecules, including IL-10, TGF-β, prostaglandin E2 (PGE2, due to the overactivation of cyclo-oxygenase II or COX-II enzyme) (Figure 1) [1][4]. These immunosuppressive molecules stimulate Th2 immune response and promote Treg polarization and function along with promoting the polarization and function of other immunosuppressive immune cells discussed later (Figure 1) [1] High-stage intraepithelial lesions are more likely to have M1 macrophages, while M2 macrophages are more common in tumors (Figure 2B) [5]. Elevated M2 levels in the TIME can create an immunosuppressive environment by supporting the generation and recruitment of myeloid-derived suppressor cells (MDSC) and regulatory T-cells (Tregs) generation and recruitment (Figure 2 and Figure 1) [1][6] while suppressing antitumor cytotoxic NK cells [7]. The increased CD163+M2 macrophages in the cervical cancer (CC) TIME are highly associated with overexpression of PD-L1 that supports immunosuppression through T-cell exhaustion and suppresses the PD-1/PD-L1 blockers’ efficacy, as discussed previously [8].
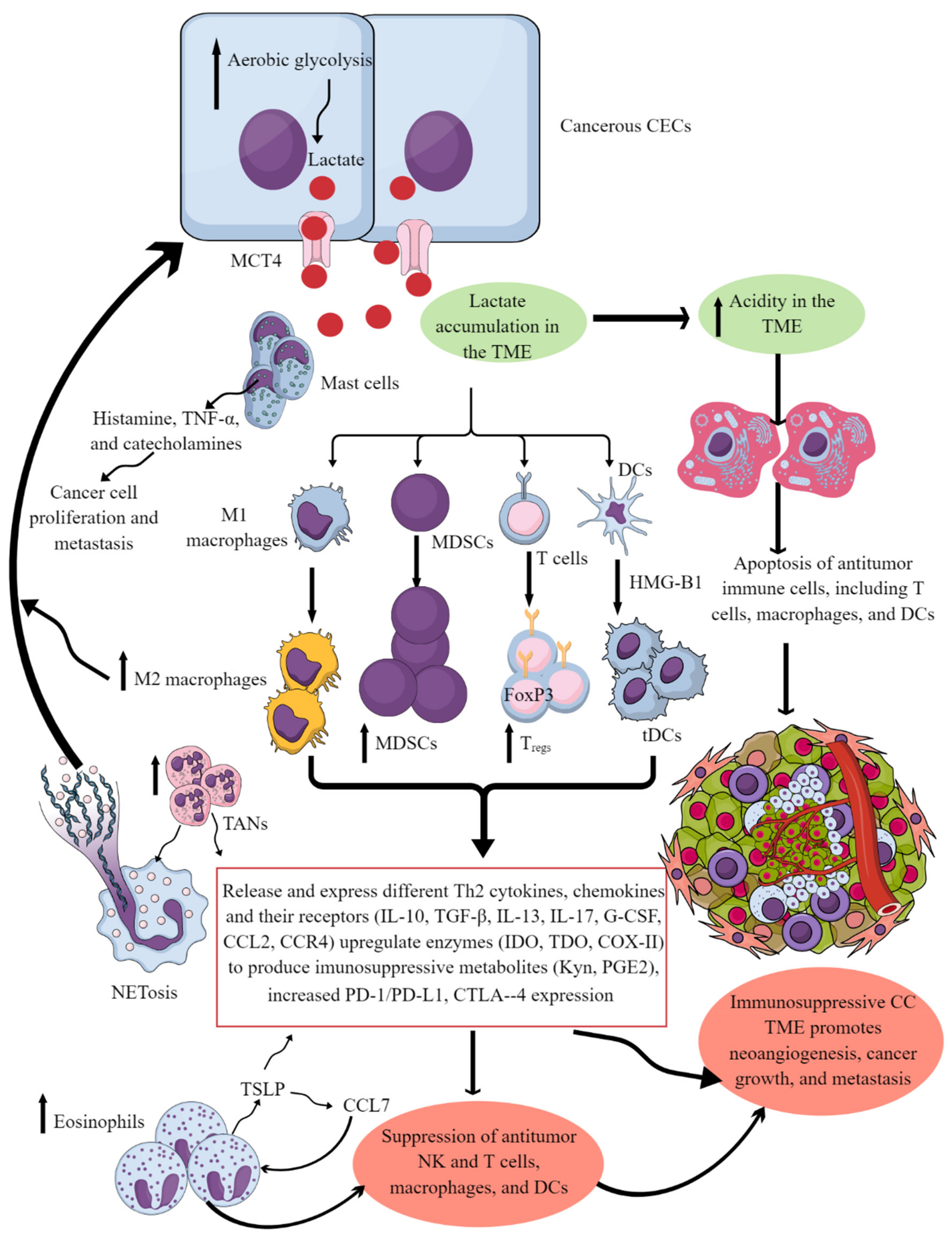
Figure 1. Cervical cancer and Immune cells crosstalk to create and support the immunosuppressive CC TIME. Cancerous CECs with increased aerobic glycolysis (due to increased energy demand to maintain their rapid growth and proliferation) overproduce lactate. This lactate is released into the TME or TIME to create an acidic microenvironment. The acidic TME induces apoptotic cell death of different antitumor immune cells and reprograms their immunometabolism to polarize them to tumor-supportive immunosuppressive immune cells (M1 to M2 macrophages, increase in MDSCs, TANs, Tregs, and tDCs). These immunosuppressive immune cells synthesize, express, and release different tumor-promoting immunosuppressive molecules for their growth and proliferation along with supporting the cancer growth, proliferation, and metastasis. These immune cells also release angiogenic factors to support neoangiogenesis for cancer survival and metastasis. Eosinophils and mast cells release several factors (TSLP, TNF-α, histamine, and catecholamines) to support immunosuppressive CC TIME and cancer-cell proliferation and metastasis. Details are mentioned in the text.
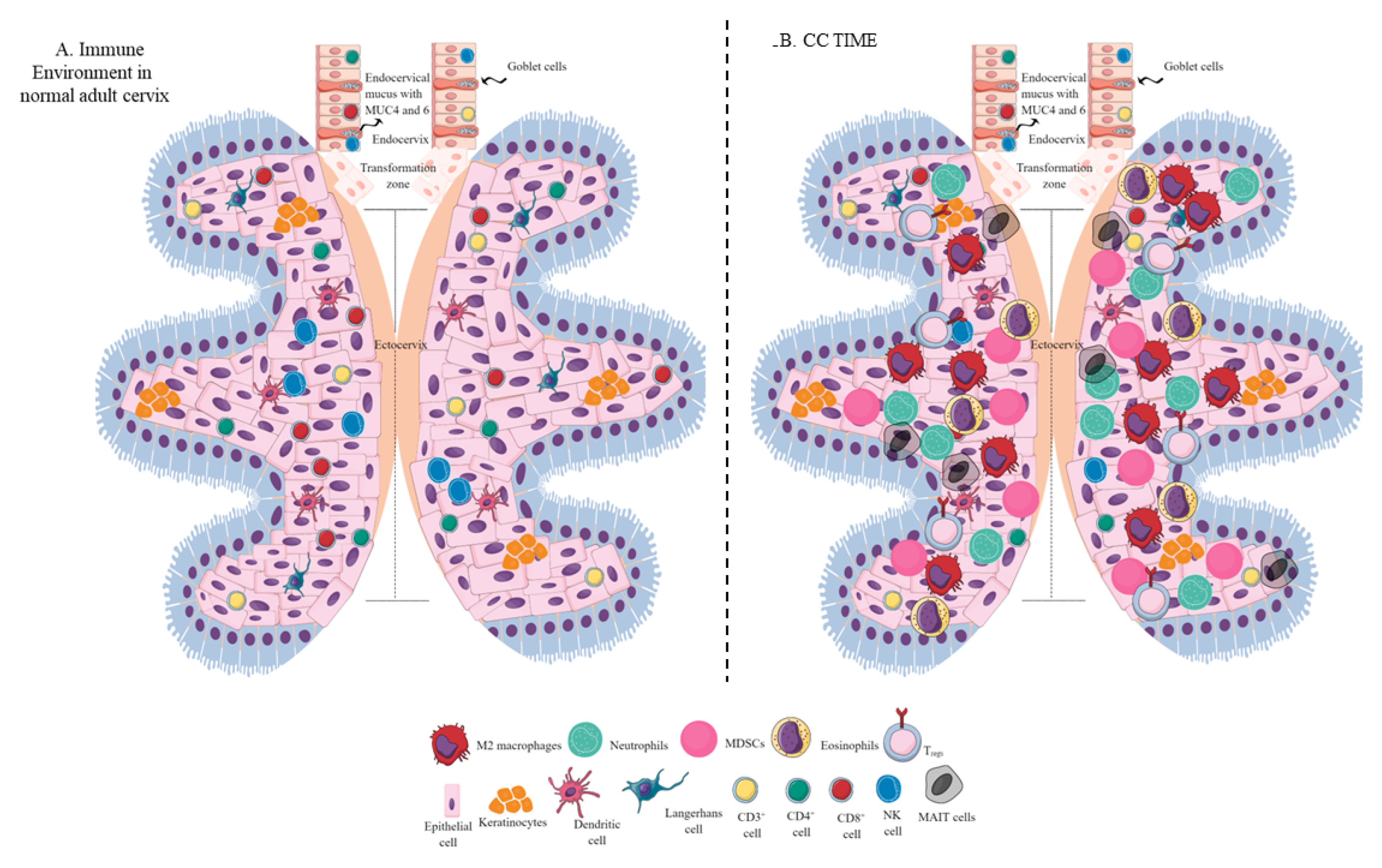
Figure 2. Schematic representation of the immune-cell population in the normal adult cervix and CC TIME. (A) Immune-cell population in the normal adult human cervix. The cervix is divided into ecto- and endocervix. Endocervix is rich in mucus and glandular goblet cells. Endocervix opens in the uterus. The zone connecting the endo- and ectocervix is called the transformation zone (TZ). Ectocervix is rich in innate immune cell population (Keratinocytes, LC, DC, macrophages, and NK cells) due to its increased chances of exposure to potential pathogens. However, the T-cell population does not vary in the endo- and ectocervix. (B). Immune-cell population in the CC TIME. CC TIME supporting tumor growth and metastasis becomes immunosuppressive and TANs, TAMs (M2 macrophages), MDSCs, tDCs, and Tregs predominate it. On the other antitumor Th1 and cytotoxic CD8+ T and NK cells decrease in number. Details are mentioned in the text.
Understanding the CC TIME can help mitigate the detrimental effects of TAMs on prognosis. For instance, blocking the expression of transmembrane protein neuropilin-1 (NRP1) in M2 macrophages can prevent M2 polarization and recruitment [9]. Additionally, radiotherapy can repolarize tumor-promoting M2 macrophages to tumor-killing M1 macrophages [10]. Mitigating M2 prevalence in the TIME could be instrumental in stimulating immune responses to target CC [11]. Developing chimeric antigen receptor (CAR)-macrophages also has excellent potential in CC immunotherapy to overcome the immunosuppressive TIME, a significant hurdle in successful cancer clearance [12][13].
2. Neutrophils in the Cervical Cancer Tumor Immune Microenvironment
Neutrophils are immune cells that can either suppress or enhance tumor growth, depending on their function in the TIME [14][15][16]. In humans, neutrophils make up 50–70% of all circulating leukocytes, but this percentage may increase in tumors [17]. An increase in neutrophil count is associated with decreased overall survival for patients with CC. Overall, CC-patient survival decreases with the increase in absolute neutrophil count (≥6187/mm3) (Figure 2B) [18]. For example, an elevated CD66b+ tumor-associated neutrophil (TAN) count is also linked to shorter recurrence-free survival in CC due to increased neutrophil extracellular traps (NETs) [19], contributing to tumor proliferation and metastasis (Figure 1) [20]. The neutrophil-mediated IL-17 release in patients with squamous CC decreases their survival by creating a tumor-promoting TIME at early tumorigenesis [21].
The neutrophil-lymphocyte ratio (NLR) can be an independent prognostic factor in CC, with an NLR ≥ 3.6 indicating a poor overall response rate and survival [22]. NLR can also help predict the CC therapeutic response. For example, patients with an NLR of less than eight have a 57% chance of one-year survival following PD-1/PD-L1 inhibitors, while individuals with an NLR of eight or below have only a 27% chance of one-year survival [23]. A possible explanation of this trend is that neutrophils are associated with elevated cytokine secretions, thus enhancing tumor growth and metastasis [22]. Thus, an overaccumulation of tumor-supportive neutrophils further supports the immunosuppressive TIME. Therefore, therapeutic exploitation of TANs is critical in the successful tumor immunotherapy [24][25][26][27].
3. Myeloid-Derived Suppressor Cells in the Cervical Cancer Tumor Immune Microenvironment
Approaches targeting MDSCs in the CC TIME are critical to overcoming the immunosuppressive TIME and increasing the efficacy of existing immunotherapies [24][25][26][27]. Three types of MDSCs including monocytic, polymorphonuclear, and early-stage cells exert potent immunosuppressive action [28][29][30]. In addition, MDSCs secrete tumor-promoting arginase that generates tumor-promoting immunosuppressive polyamines in the CC TIME [28][31]. However, MDSCs activity in CC is governed by many aspects of the TIME, including immune and nonimmune cells and released factors. For example, growth factor granulocyte-colony stimulating factor (G-CSF) increases MDSCs’ number in CC (Figure 2B and Figure 1). Elevated G-CSF can induce tumor-related leukocytosis (TRL), a condition found in patients with advanced cancer [32]. Specifically, patients with CC with TRL are at greater risk for metastasis and are low responders to radiotherapy [33][34]. Given the connection and detrimental effect of MDSCs and G-CSF levels, future CC therapies should focus on alternative therapies to mitigate these levels. Novel immunotherapeutic approaches could focus on mitigating MDSCs and controlling G-CSF levels.
Furthermore, T cells also affect MDSCs’ function in TIME and vice-versa [35]. The combined activity of m-MDSCs and mucosal-associated invariant T (MAIT)-cells may be associated with CC progression (Figure 2B) [36]. All-trans retinoic acid (ATRA, a vitamin A-derivative) influences MDSCs maturation and eliminates their immunosuppressive activity. ATRA treatment decreases MDSC accumulation in BALB/C mice with CC and increases antitumor cytotoxic CD8+ T cells [37]. Combined ATRA and anti-PD-L1 therapies may be promising approaches to CC cancer immunotherapy, as this approach delays tumor growth and increases antitumor T-cells, IFN-γ, and TNF-α levels [37]. Thus, approaches targeting MDSCs in the CC TIME are critical to overcoming the immunosuppressive TIME and increasing the efficacy of existing immune checkpoint inhibitors (ICIs) and other immunotherapies.
4. Mucosal-Associated Invariant T Cells in the Cervical Cancer Tumor Immune Microenvironment
Mucosal-associated invariant T (MAIT) cells are unique T cells found in the body’s peripheral blood, liver, and mucosal surfaces, including the cervix [38]. They produce cytokines such as IFN-γ and IL-17, which are crucial in fighting pathogens such as bacteria, viruses, and fungi. They also regulate inflammatory responses and contribute to immune-mediated diseases [38][39][40][41]. MAIT cells are essential in different cancers and have potential in immunotherapy [42][43][44][45][46][47][48].
In patients with CC, there is a decrease in the number of MAIT cells in circulation, which is associated with poor progression-free survival [49]. The number of CD4-CD8-PD1+ MAIT (DN or double negative) cells in the peripheral circulation of patients with CC is directly related to disease severity [36]. The decrease in peripheral DN MAIT cells indicates that they might have migrated to the cancer tissue, further supporting the growth of the tumor [36]. Further investigation into the number and types of MAIT cells in CC biopsies and animal studies is necessary to understand this critical area of tumor immunology.
MAIT cells in the tumor microenvironment (TME) can either hinder or aid the antitumor activity of NK cells. While MAIT cell accumulation can contribute to tumor growth and spread, activating these cells can enhance their antitumor action by activating NK cells [50]. In cancer immunotherapy, inhibiting MHC class I-related protein 1 (MR-1) on cancer cells can be used to develop MAIT-cells-based treatments. The interaction with MR-1 expressed on tumor cells and TIME MAIT cells activate them to release IL-17A, suppressing cytotoxic T and NK cells. Thus, blocking MR-1 on cancer cells may help design MAIT-cells-based cancer immunotherapy. Additionally, combining a synthetic riboflavin synthesis pathway-derived antigen 5-OP-RU [5-(2-oxopropylideneamino)-6-D-ribitylaminouracil] and the CpG (a TLR9 agonist) can boost the antitumor immune response of TIME MAIT cells as indicated by the increased levels of CD69 expression, pronounced effector memory phenotype, and upregulation of effector molecules, including IFN-γ, granzyme B (GrB), and perforin [51]. Interestingly, 5-OP-RU and TLR9 agonist combination work independently of MHC class I related-1 molecule (MR1) expression in tumor cells. Reprogramming and redifferentiating TIME MAIT cells are also promising approaches for cancer immunotherapy. Therefore, exploring and targeting TIME MAIT cells in CC is a new and innovative cellular immunotherapy strategy.
5. Mast Cells in the Cervical Cancer Tumor Immune Microenvironment
Mast cells are critical innate immune cells with different immune and inflammation regulatory functions [52][53]. Initially, they were only associated with allergic reactions/diseases. However, recent advancements in immunology have established them as potent immunoregulatory cells that perform various immune functions, including maintaining immune homeostasis [52][53][54][55]. Mast cells play a critical role in the TIME of many cancers and their effect on promoting or inhibiting tumors depends on the type and stage of cancer [56][57]. Mast cells (tryptase-positive and tryptase/chymase-positive) are also found in the normal human cervix, and their number increases in benign inflammatory conditions [58][59][60]. A recent study reported a widespread distribution of mast cells in CC tissues and patients with low mast-cell density in their TIME had better overall survival rates [61]. Mast cells promote tumor growth by supporting neoangiogenesis and creating an immunosuppressive TIME. Specific mast-cell mediators, including histamine, TNF-α, and cannabinoids, also contribute to CC cell invasion and metastasis (Figure 3) [54][55][56][57][62]. In addition, mast-cell infiltration in the TIME increases the tumor’s resistance to anti-PD-1 immune checkpoint blockers [63][64]. Hence, targeting mast cells and their mediators in the CC TIME may inhibit CC growth and metastasis by inhibiting neoangiogenesis and immunosuppressive events that support CC metastasis.
6. Eosinophils in the Cervical Cancer Tumor Immune Microenvironment
Eosinophils are critical innate immune cells that are present in low numbers in blood but are present in higher numbers at mucosal surfaces [65][66][67]. They play a critical role in antimicrobial immunity, allergies, and tumor immunity [68][69][70]. For example, eosinophils work with DCs and T cells to produce inflammatory and adaptive immune responses [71]. A patient’s eosinophil count can predict the immune response in cervical SCC, and high levels of eosinophils can result in poor survival (Figure 2B) [72][73]. Hypoxic conditions influence the function of EOs during cancer progression. The prevalence of eosinophils increases with CC progression, and hypoxic conditions also influence their function [64][74]. Thymic stromal lymphopoietin (TSLP) stimulates CC growth [75] and regulates eosinophil activity in the hypoxic CC TME. The increased TSLP upregulates CCL17 production [64][75], which over-recruits eosinophils (Figure 1). In addition, TSLP promotes CC progression by promoting the immunosuppressive Th2 immune response (Figure 1). Eosinophils can create an immunosuppressive TIME by releasing Th2 cytokines and suppressing NK and T-cell functions in the CC (Figure 1) [76][77].
7. Drug Conjugates in the Cervical Cancer Tumor Immune Microenvironment
Drug conjugates (DCs) are an essential immune system component that can either enhance or suppress tumor response [71][72][73][74][75][76][77][78]. The different types of DCs, including conventional DCs (cDC1s and cDC2s), plasmacytoid DCs (pDCs), and mature DCs, express varying levels of costimulatory molecules and immune checkpoints, depending on the type and stage of the tumor [79][80]. For instance, cDC1s typically do not express PD-L1 and immunoglobulin-like transcript 2 (ILT2) under normal conditions. However, during tumor progression, they may express high T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), a unique immune-checkpoint repertoire [80]. TIM-3 interacts with galectin-9, a C-type lectin, to stimulate antitumor function in innate immune cells, such as DCs, NK cells, and macrophages, by activating proinflammatory signaling pathways, including PI3K-mammalian target of rapamycin (mTOR) and hypoxia-inducible factor-1 (HIF-1) signaling without inducing apoptosis [74][75][76][77][78][79].
The decrease of galectin-9 in CC (CIN and SCC) patients increases its severity, whereas its increase is associated with a better prognosis regarding the overall survival [80][81]. However, severe CC cases (advanced stage IV) have an increased systemic galectin-9 level, indicating that circulating galectin-9 via TIM-3 interaction in systemic Th1 and CD8+ T cells induces their apoptosis and impairs their infiltration in the CC TME [82]. Furthermore, in HPV-associated patients with CC, circulating CD4+ and CD8+ T cells overexpress TIM-3, supporting that increased circulating galectin-9, including monocyte-specific galectin-9 in patients with CC, induces Th1 and CD8+ T cell apoptosis to suppress systemic T-cell-dependent antitumor immunity [83]. Thus, increased circulating TIM-3 expression of T cells and galectin-9 in patients with CC is associated with poor CC prognosis. In addition, increased TIM-3 expression in Tregs via galectin-9 interaction increases the immunosuppressive function (IL-10 and TGF-β release) in patients with CC [83][84]. Notably, the systemic galectin-9 level is independent of the local CC TME. Hence, local CC TME galectin-9 decreases the CC severity and improves overall patient survival, whereas systemic galectin-9 is associated with increased CC severity. Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1) is an adhesion molecule that also serves as a heterophilic ligand for TIM-3 expressed on T cells [85]. In severe or high-grade squamous intraepithelial lesions (SIL) of patients with CC, the increased CEACAM-1 and TIM-3 interaction further suppresses the antitumor activity of CD4+ and CD8+ T cells [86]. Furthermore, CC cells overexpress HMGB1 and serve as a prognostic indicator and a potential biomarker, suppressing antitumor T-cell function via interacting with TIM-3 [87][88][89][90][91].
cDC1s are critical for antitumor cytotoxic T cells and decrease Tregs via secreting the cGAS/STING signaling pathway-dependent type 1 IFNs [92]. The cDC1s’ decrease in the CAC TIME is associated with poor patient survival due to impaired T-cell-mediated antitumor immunity (Figure 2B and Figure 1) [93]. Although cDC1s highly express TIM-3, their decrease in severe CC cases indicates that they could not exert their antitumor action. Therefore, it should be interesting to investigate whether they die or transform to tumor-supportive tolerogenic DCs in the immunosuppressive CC TIME. The increased HMGB1 level in the CC TIME is associated with promoting pDCs to tDCs that further supports immunosuppressive TIME for the CC progression (Figure 3 and Figure 1) [94]. Furthermore, low cDC1 chemo-attractive chemokines in the CC TIME support the immunosuppressive niche. For example, NK cells in the TIME support cDC1 infiltration by releasing XCL1 and CCL5, and NK cells lose this function in the presence of PGE2 [95]. Overexpressed PGE2 in the CC TIME suppresses NK cell-mediated release of cDC1 chemokines to dysregulate the NK cell–cDC1–chemokine axis [96][97]. Thus, HMGB1 and PGE2 in the CC TIME suppress antitumor T cell, NK cell, and DC function, supporting CC growth and metastasis (Figure 1).
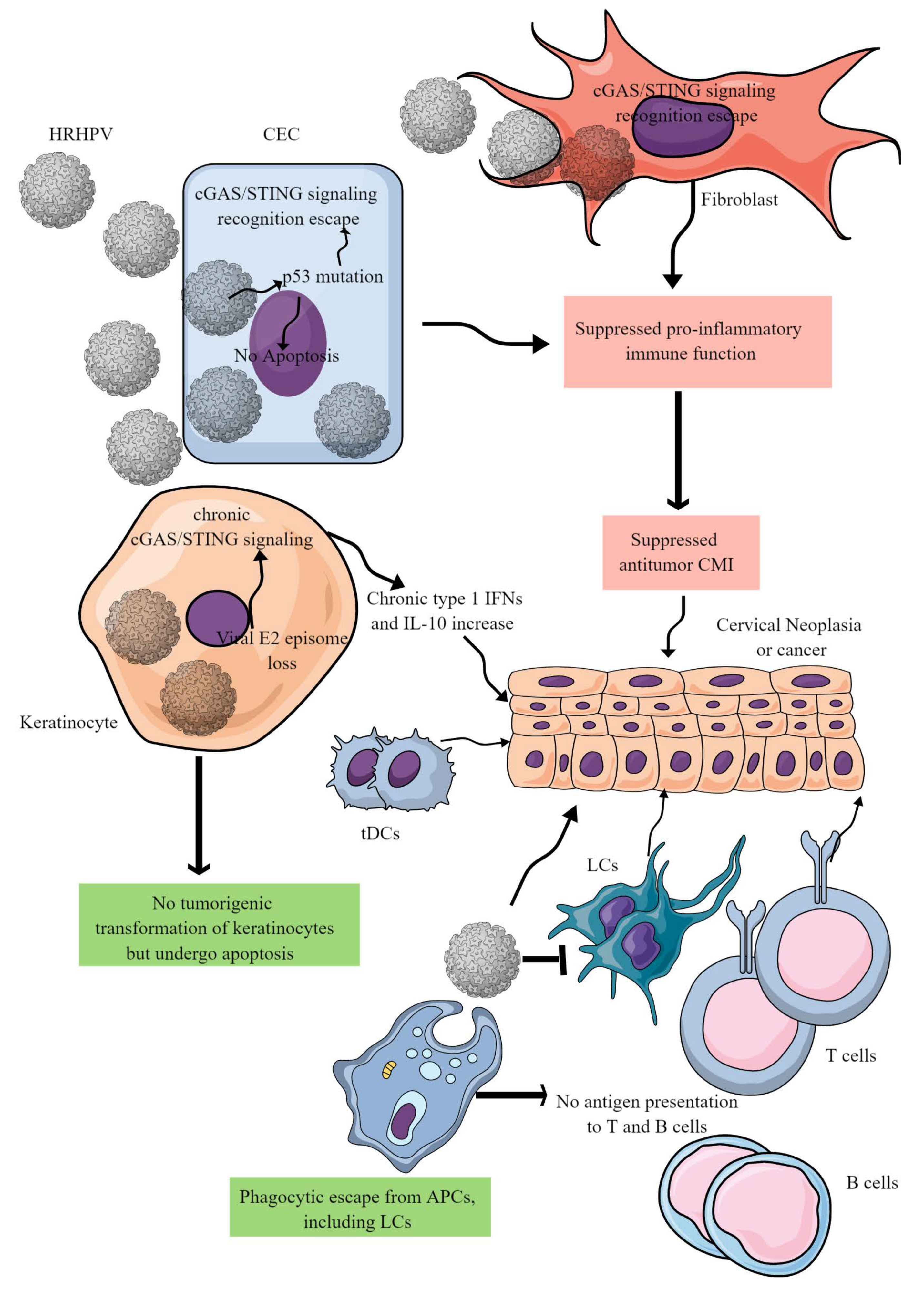
Figure 3. HR-HPV-mediated CC immunopathogenesis. HR-HPVs infect basal epithelial cells, keratinocytes, and fibroblasts. HR-HPV infection transforms epithelial cells into neoplastic cells and later into cancer. The other keratinocytes escape from tumorigenic transformation and undergo apoptosis to support the immunosuppressive environment for tumor growth. In epithelial cells, HR-HPV infection prevents their apoptosis by inducing p53 mutation that also inhibits the cGAS/STING-mediated antitumor immune response. In addition, other proinflammatory innate immune functions of epithelial cells are also blocked, creating a chronic tumor-supportive immunosuppressive niche. Notably, in keratinocytes, only cGAS/STING-mediated NF-κB-mediated release of proinflammatory cytokines is inhibited leaving the type 1 IFN generation intact. This leads to the chronic type 1 IFN generation, which supports tumor growth. Furthermore, HR-HPV escapes phagocytosis by antigen-presenting cells (APCs, macrophages, LCs, and DCs) and antigen presentation to T and B cells. Metabolites released in the TIME by cancer cells further suppress cDC1s and NK cells. For example, DCs repolarize to tDCs in the presence of tryptophan and its metabolism by IDO. Details are discussed in the text.
HPV-VLP vaccination stimulates DC and NK cell crosstalk to exert the antitumor activity in patients with CC, as indicated by the CD69 (an activation marker) and HLA-DR upregulation on DCs and increased NKCC and IFN-γ release [98][99][100]. DC-derived exosome vaccines also induce antitumor cytotoxic CD8+ T cell activity, proliferation, and IFN-γ secretion [101]. Immunotherapeutics work effectively under the right immunological conditions. For example, the synthetic dsRNA viral analog Poly I: C (polyinosinic: polycytidylic acid) vaccine is a promising CC vaccine. Poly I: C vaccine induces receptor-interacting protein kinase 3 (RIPK3) signaling for its direct cytotoxicity on tumor cells. The CC-cell necroptosis induces IL-1α release, which activates DC-mediated IL-12 production, critical for an antitumor immune response [102]. Thus, increasing cDC1s in CAC patients will increase their survival via increasing antitumor immunity. Further studies in this direction are critical to designing DC-specific vaccines for CC.
8. Natural Killer Cells in the Tumor Immune Microenvironment
NK cells are potent antitumor innate immune cells categorized as type 1 innate lymphoid cells (ILCs) [81]. Their role in antitumor immunity, including uterine cancer, has been discussed elsewhere [82][83]. However, in CC, the number and function of NK cells are reduced due to various cellular processes (Figure 2B). For example, CD3+CD56+ NK cell infiltration increases at early CC stages, which decreases as cancer progresses to advanced stages due to higher TGF-β1 in the tumor. TGF-β1 inhibits natural-killer group 2D (NKG2D), CD16, and Ki67 receptor function [84]. The decreased NK cell number in CC TIME is associated with HLA-I downregulation, potentially due to upregulated immunosuppressive cytokines, including IL-10, IL-13, and TGF-β [85], which inhibit NK-cell function. Patients with CC have HLA-E (a major histocompatibility (MHC) class I molecules involved in the NK-cell recognition pathway) overexpression, which is not well-associated with the prognostic outcome, potentially due to a high volume of exhausted or apoptotic CD8+ T cells [86]. However, elevated HLA-E expression in patients with CAC improves survival [87]. The decreased NK-cell number in the CC TIME further supports the decrease in potent antitumor cDC1s in patients with advanced CC. Therefore, the strategies to design NK-cell-based immunotherapies to target CC will be an exciting area to explore.
9. T Cells in the Tumor Immune Microenvironment
The type and concentration of T-cells in a person’s immune system can provide insight into their immune response and prognosis for CC. T cells infiltrate CC tumors, but the CD4+: CD8+ differs from that in the peripheral blood [88] and lower CD4/CD8 ratios are associated with faster tumor growth and lymph-node metastasis [89]. For example, healthy women have a CD4/CD8 ratio of 1.42 [90] but this number decreases to 0.6 and 1.17 in women with fatal and nonfatal CC, respectively [91]. These trends can be explained mechanistically, as CD4+ T cells activate the cytotoxic CD8+ T cells, and Tregs accumulate near advanced tumors, inhibiting antitumor immune activity. Th1 cells’ number and function also alter CC carcinogenesis (Figure 2B). For example, Th1 levels increase from low to high-grade squamous intraepithelial lesions but deplete from high-grade squamous intraepithelial lesions to SCC. In contrast, Th2 levels deplete from low- to high-grade squamous intraepithelial lesions [92]. Th2 and Th17 populations increase, and Th1 levels are depleted in CIN and CC, which supports that a shift in these cell populations starts prior to CC formation and, thus, contributes to CIN progressing into CC (Figure 2B) [93]. Th1 dominance is critical to antitumor immunity and contributes to immune memory, forming tumor-specific cytotoxic T-lymphocytes (CTLs) [94], further suggesting that Th1 depletion is crucial to CIN progression into CC.
HPV infections preceding CC development contribute to T-cell alterations. For example, CD4+ T-cells specific to HPV+ patients with CC can suppress T-cell proliferation and alter their function [94]. The ratio of cell types changes not only with cancer status but also with HPV status. For example, CD8+ T cells are more prevalent than CD4+ in the epithelial layer of an HPV+ normal cervix, but this becomes less prominent with an increasing CIN grade [96]. The CD4/CD8 ratio, as well as the quantity of CD4+ T cells, are indicative of CC survival in HPV+ individuals. Overall, a lower number of Tregs is detrimental to five-year survival but, more specifically, individuals with lower CD4/CD8 ratios have higher mortality rates than those with higher ratios [91]. Women undergoing neoadjuvant chemotherapy have more remarkable survival if they have higher CD4/CD8 ratios before their third round of treatment [97]. Furthermore, neoadjuvant chemotherapy increases CD4, CD8, CD20, and CD56 signals, most prominently in good responders, indicating the activation of antitumor Th1, cytotoxic T, B, and NK cells [98]. Therefore, the immunoactive TIME in good responders is crucial to support locoregional stimulation of antitumor immunity during neoadjuvant chemotherapy. Hence, neoadjuvant chemotherapy can be combined with ICIs in patients with CC to stimulate antitumor immunity.
Specific gene-expression profiles and ligands can significantly impact Tregs within the CC TIME. For example, Foxp3 and V-domain immunoglobulin suppressor of T-cell activation (VISTA) significantly correlate with CC prognosis, exhibiting higher expression in CC than in CIN or chronic cervicitis. Specifically, patients with double-negative (Foxp3 and VISTA) tumors show the best prognosis, while double-positive patients show the worst prognosis [99]. Foxp3 levels are also higher in patients with lymph node metastasis than those without metastasis [100]. Foxp3 levels are also higher in patients with CC with lymph-node metastasis than those without metastasis [100]. The increased FoxP3 expression in Th1 cells to transform them to Tregs occurs due to the intracellular STING activation in these T cells [101]. The intrinsic STING activation in T cells induces TANK binding kinase-1-interferon regulatory factor (TBK1-IRF3)-mediated mothers against decapentaplegic homolog 3 or SMAD3 and signal transducer and activator of transcription 5 (STAT5) phosphorylation independent of IFN-β to induce FoxP3 activation and their transformation to Tregs. In CC TIME, tumor-derived exosomes with TGF-β, cGAS, and 2′-3′-cGAMP activate STING signaling in tumor-infiltrated T cells to promote induced-Treg (iTreg) expansion [101].
Understanding T-cell-specific TIL activity changes is critical to designing better T-cell-based immunotherapeutics specific to CC type. Patients with CC have better effector T-cell infiltration than adenocarcinoma patients, with elevated CD45+ and CD3+ levels, Tregs, and PD-1 and TIM-3 immune checkpoints. These changes are prognostically significant and may indicate immunotherapeutic responses, as increases in CD3+ densities can decrease the death and relapse risk [102]. Therefore, immunotherapeutics that work well for SCC may not work as well in adenocarcinoma. Nevertheless, tumor-specific T cells are ideal candidates for personalized, adaptive immunotherapy. TILs from individual patients are primed for specific tumors and are immediately ready to return after infusion [103].
Under conditions that support oxidative phosphorylation (OXPHOS), Th17 cells have increased persistence and can decrease tumor growth in vivo [104]. In squamous CC TIME, an increased presence of Th17 cells has been associated with improved patient survival due to their anticancer effects [21][105]. The decreased presence of galectin-9 in the CC TIME promotes the development of Th17 cells, as galectin-9 interaction with TIM-3 induces apoptosis in mature Th17 cells [106]. However, Th17 cells in CAC are detrimental to the patient and can increase tumor growth and severity and contribute to CAC relapse after tumor removal [107]. The infiltration of Th17 cells in the CC TIME is facilitated by CCL20, which binds to overexpressed CCR6 [108]. Therefore, it would be interesting to explore the immunometabolic reprogramming of Th17 cells that govern their anti- and protumor functions in squamous and adenocarcinoma patients with CC to develop immunometabolism-based Th17 cell-directed immunotherapies.
10. B Cells in the Tumor Immune Microenvironment
B-cells play a crucial role in regulating the immune system by producing antibodies and releasing cytokines [109]. Studies on mice have shown that reducing the B-cell count can boost the body’s antitumor response by lowering IL-10 production and increasing IFN-γ levels from CD8+ T-cells and NK cells [110]. However, a subset of B-cells called Bregs can hinder cytokine secretion and counteract the antitumor response of other immune cells [111]. Understanding the role of B-cells in cancer immunity is critical, especially for HPV-associated cancers, which have shown conflicting results in human patients. For example, B-cells exert a vital antitumor role in HPV+ patients with CC [112]. Therefore, a deeper understanding of a patient’s cancer type is required in treatment.
Researchers are exploring the potential of B-cell-targeted immunotherapy in cancer treatment. For instance, PD-1 blockade and radiotherapy have proven effective in increasing memory B-cells, antigen-specific B-cells, and plasma cells in HPV-associated cancers [112]. However, Bregs have been found to inhibit CD8+ T-cell cytotoxicity in CC, leading to lower prognostic outcomes in individuals with low CD4/CD8 ratios [113]. There is still much to learn about the role of B-cells and Bregs in CC and their modulation of immunotherapy.
This entry is adapted from the peer-reviewed paper 10.3390/biology12070941
References
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239.
- Ma, R.-Y.; Black, A.; Qian, B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022, 43, 546–563.
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In Macrophage Activation; Khalid Hussain, B., Ed.; IntechOpen: Rijeka, Croatia, 2019; Chapter 1.
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389.
- Li, C.; Hua, K. Dissecting the Single-Cell Transcriptome Network of Immune Environment Underlying Cervical Premalignant Lesion, Cervical Cancer and Metastatic Lymph Nodes. Front. Immunol. 2022, 13, 897366.
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug. Discov. 2022, 21, 799–820.
- Nuñez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Raffo Iraolagoitia, X.L.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-β and Engagement of CD85j. J. Immunol. 2018, 200, 1008–1015.
- Guo, F.; Feng, Y.C.; Zhao, G.; Zhang, R.; Cheng, Z.Z.; Kong, W.N.; Wu, H.L.; Xu, B.; Lv, X.; Ma, X.M. Tumor-Associated CD163(+) M2 Macrophage Infiltration is Highly Associated with PD-L1 Expression in Cervical Cancer. Cancer Manag. Res. 2020, 12, 5831–5843.
- Chen, X.-J.; Wu, S.; Yan, R.-M.; Fan, L.-S.; Yu, L.; Zhang, Y.-M.; Wei, W.-F.; Zhou, C.-F.; Wu, X.-G.; Zhong, M.; et al. The role of the hypoxia-Nrp-1 axis in the activation of M2-like tumor-associated macrophages in the tumor microenvironment of cervical cancer. Mol. Carcinog. 2019, 58, 388–397.
- Ren, J.; Li, L.; Yu, B.; Xu, E.; Sun, N.; Li, X.; Xing, Z.; Han, X.; Cui, Y.; Wang, X.; et al. Extracellular vesicles mediated proinflammatory macrophage phenotype induced by radiotherapy in cervical cancer. BMC Cancer 2022, 22, 88.
- Krneta, T.; Gillgrass, A.; Poznanski, S.; Chew, M.; Lee, A.J.; Kolb, M.; Ashkar, A.A. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 2017, 101, 285–295.
- Wang, S.; Yang, Y.; Ma, P.; Zha, Y.; Zhang, J.; Lei, A.; Li, N. CAR-macrophage: An extensive immune enhancer to fight cancer. eBioMedicine 2022, 76, 103873.
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953.
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620.
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187.
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503.
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446.
- Nomelini, R.S.; Mota, S.D.S.; Murta, E.F.C. Absolute band neutrophils count is a predictor of overall survival in advanced uterine cervical cancer. Arch. Gynecol. Obstet. 2022, 306, 1697–1701.
- Carus, A.; Ladekarl, M.; Hager, H.; Nedergaard, B.S.; Donskov, F. Tumour-associated CD66b+ neutrophil count is an independent prognostic factor for recurrence in localised cervical cancer. Br. J. Cancer 2013, 108, 2116–2122.
- Yan, B.; Dai, X.; Ma, Q.; Wu, X. Stromal Neutrophil Extracellular Trap Density Is an Independent Prognostic Factor for Cervical Cancer Recurrence. Front. Oncol. 2021, 11, 659445.
- Punt, S.; Fleuren, G.J.; Kritikou, E.; Lubberts, E.; Trimbos, J.B.; Jordanova, E.S.; Gorter, A. Angels and demons: Th17 cells represent a beneficial response, while neutrophil IL-17 is associated with poor prognosis in squamous cervical cancer. Oncoimmunology 2015, 4, e984539.
- Ittiamornlert, P.; Ruengkhachorn, I. Neutrophil-lymphocyte ratio as a predictor of oncologic outcomes in stage IVB, persistent, or recurrent cervical cancer patients treated by chemotherapy. BMC Cancer 2019, 19, 51.
- Calo, C.A.; Barrington, D.A.; Brown, M.; Gonzalez, L.; Baek, J.; Huffman, A.; Benedict, J.; Backes, F.; Chambers, L.; Cohn, D.; et al. High pre-treatment neutrophil-to-lymphocyte ratio as a prognostic marker for worse survival in patients with recurrent/metastatic cervical cancer treated with immune checkpoint inhibitors. Gynecol. Oncol. Rep. 2022, 42, 101040.
- Gruijs, M.; Sewnath, C.A.N.; van Egmond, M. Therapeutic exploitation of neutrophils to fight cancer. Semin. Immunol. 2021, 57, 101581.
- Carnevale, S.; Ghasemi, S.; Rigatelli, A.; Jaillon, S. The complexity of neutrophils in health and disease: Focus on cancer. Semin. Immunol. 2020, 48, 101409.
- Geh, D.; Leslie, J.; Rumney, R.; Reeves, H.L.; Bird, T.G.; Mann, D.A. Neutrophils as potential therapeutic targets in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 257–273.
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275.
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25.
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220.
- Raskov, H.; Orhan, A.; Gaggar, S.; Gögenur, I. Neutrophils and polymorphonuclear myeloid-derived suppressor cells: An emerging battleground in cancer therapy. Oncogenesis 2022, 11, 22.
- Wesolowski, R.; Markowitz, J.; Carson, W.E. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunotherapy Cancer 2013, 1, 10.
- Kawano, M.; Mabuchi, S.; Matsumoto, Y.; Sasano, T.; Takahashi, R.; Kuroda, H.; Kozasa, K.; Hashimoto, K.; Isobe, A.; Sawada, K.; et al. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci. Rep. 2015, 5, 18217.
- Sasano, T.; Mabuchi, S.; Kozasa, K.; Kuroda, H.; Kawano, M.; Takahashi, R.; Komura, N.; Yokoi, E.; Matsumoto, Y.; Hashimoto, K.; et al. The Highly Metastatic Nature of Uterine Cervical/Endometrial Cancer Displaying Tumor-Related Leukocytosis: Clinical and Preclinical Investigations. Clin. Cancer Res. 2018, 24, 4018–4029.
- Cho, Y.; Kim, K.H.; Yoon, H.I.; Kim, G.E.; Kim, Y.B. Tumor-related leukocytosis is associated with poor radiation response and clinical outcome in uterine cervical cancer patients. Ann. Oncol. 2016, 27, 2067–2074.
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J. Immunol. 2013, 191, 17–23.
- Lu, Z.; Zhu, M.; Marley, J.L.; Bi, K.; Wang, K.; Zhai, M.; Hu, H.; Guo, P.; Li, C.; Xu, Y.; et al. The combined action of monocytic myeloid-derived suppressor cells and mucosal-associated invariant T cells promotes the progression of cervical cancer. Int. J. Cancer 2021, 148, 1499–1507.
- Liang, Y.; Wang, W.; Zhu, X.; Yu, M.; Zhou, C. Inhibition of myeloid-derived suppressive cell function with all-trans retinoic acid enhanced anti-PD-L1 efficacy in cervical cancer. Sci. Rep. 2022, 12, 9619.
- Kumar, V.; Ahmad, A. Role of MAIT cells in the immunopathogenesis of inflammatory diseases: New players in old game. Int. Rev. Immunol. 2018, 37, 90–110.
- Ussher, J.E.; Willberg, C.B.; Klenerman, P. MAIT cells and viruses. Immunol. Cell Biol. 2018, 96, 630–641.
- Rouxel, O.; Lehuen, A. Mucosal-associated invariant T cells in autoimmune and immune-mediated diseases. Immunol. Cell Biol. 2018, 96, 618–629.
- Hinks, T.S.C.; van Wilgenburg, B.; Wang, H.; Loh, L.; Koutsakos, M.; Kedzierska, K.; Corbett, A.J.; Chen, Z. Study of MAIT Cell Activation in Viral Infections In Vivo. Methods Mol. Biol. 2020, 2098, 261–281.
- Zumwalde, N.A.; Gumperz, J.E. Mucosal-Associated Invariant T Cells in Tumors of Epithelial Origin. Adv. Exp. Med. Biol. 2020, 1224, 63–77.
- Berzins, S.P.; Wallace, M.E.; Kannourakis, G.; Kelly, J. A Role for MAIT Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 949.
- Shaler, C.R.; Tun-Abraham, M.E.; Skaro, A.I.; Khazaie, K.; Corbett, A.J.; Mele, T.; Hernandez-Alejandro, R.; Haeryfar, S.M.M. Mucosa-associated invariant T cells infiltrate hepatic metastases in patients with colorectal carcinoma but are rendered dysfunctional within and adjacent to tumor microenvironment. Cancer Immunol. Immunother. 2017, 66, 1563–1575.
- Haeryfar, S.M.M.; Shaler, C.R.; Rudak, P.T. Mucosa-associated invariant T cells in malignancies: A faithful friend or formidable foe? Cancer Immunol. Immunother. 2018, 67, 1885–1896.
- Melo, A.M.; O’Brien, A.M.; Phelan, J.J.; Kennedy, S.A.; Wood, N.A.W.; Veerapen, N.; Besra, G.S.; Clarke, N.E.; Foley, E.K.; Ravi, A.; et al. Mucosal-Associated Invariant T Cells Display Diminished Effector Capacity in Oesophageal Adenocarcinoma. Front. Immunol. 2019, 10, 1580.
- O’Neill, C.; Cassidy, F.C.; O’Shea, D.; Hogan, A.E. Mucosal Associated Invariant T Cells in Cancer-Friend or Foe? Cancers 2021, 13, 1582.
- Li, Y.-R.; Zhou, K.; Wilson, M.; Kramer, A.; Zhu, Y.; Dawson, N.; Yang, L. Mucosal-associated invariant T cells for cancer immunotherapy. Mol. Ther. 2023, 31, 631–646.
- Huang, W.-C.; Hsiao, Y.-C.; Wu, C.-C.; Hsu, Y.-T.; Chang, C.-L. Less circulating mucosal-associated invariant T cells in patients with cervical cancer. Taiwan. J. Obstet. Gynecol. 2019, 58, 117–121.
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141.
- Ruf, B.; Catania, V.V.; Wabitsch, S.; Ma, C.; Diggs, L.P.; Zhang, Q.; Heinrich, B.; Subramanyam, V.; Cui, L.L.; Pouzolles, M.; et al. Activating Mucosal-Associated Invariant T Cells Induces a Broad Antitumor Response. Cancer Immunol. Res. 2021, 9, 1024–1034.
- Kolkhir, P.; Elieh-Ali-Komi, D.; Metz, M.; Siebenhaar, F.; Maurer, M. Understanding human mast cells: Lesson from therapies for allergic and non-allergic diseases. Nat. Rev. Immunol. 2022, 22, 294–308.
- Kumar, V.; Sharma, A. Mast cells: Emerging sentinel innate immune cells with diverse role in immunity. Mol. Immunol. 2010, 48, 14–25.
- Abraham, S.N.; St John, A.L. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010, 10, 440–452.
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142.
- Aponte-López, A.; Muñoz-Cruz, S. Mast Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 159–173.
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325.
- Cabanillas-Saez, A.; Schalper, J.A.; Nicovani, S.M.; Rudolph, M.I. Characterization of mast cells according to their content of tryptase and chymase in normal and neoplastic human uterine cervix. Int. J. Gynecol. Cancer 2002, 12, 92–98.
- Naik, R.; Pai, M.R.; Poornima Baliga, B.; Nayak, K.S.; Shankarnarayana; Dighe, P. Mast cell profile in uterine cervix. Indian J. Pathol. Microbiol. 2004, 47, 178–180.
- Kalyani, R.; Rajeshwari, G. Significance of mast cells in non-neoplastic and neoplastic lesions of uterine cervix. Biomed. Res. Ther. 2016, 3, 3.
- Guo, F.; Kong, W.N.; Li, D.W.; Zhao, G.; Wu, H.L.; Anwar, M.; Shang, X.Q.; Sun, Q.N.; Ma, C.L.; Ma, X.M. Low Tumor Infiltrating Mast Cell Density Reveals Prognostic Benefit in Cervical Carcinoma. Technol. Cancer Res. Treat. 2022, 21, 15330338221106530.
- Benítez-Bribiesca, L.; Wong, A.; Utrera, D.; Castellanos, E. The role of mast cell tryptase in neoangiogenesis of premalignant and malignant lesions of the uterine cervix. J. Histochem. Cytochem. 2001, 49, 1061–1062.
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346.
- Li, J.; Peng, G.; Zhu, K.; Jie, X.; Xu, Y.; Rao, X.; Xu, Y.; Chen, Y.; Xing, B.; Wu, G.; et al. PD-1+ mast cell enhanced by PD-1 blocking therapy associated with resistance to immunotherapy. Cancer Immunol. Immunother. 2023, 72, 633–645.
- Shamri, R.; Xenakis, J.J.; Spencer, L.A. Eosinophils in innate immunity: An evolving story. Cell Tissue Res. 2011, 343, 57–83.
- Travers, J.; Rothenberg, M.E. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015, 8, 464–475.
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174.
- Kita, H. Eosinophils: Multifaceted biological properties and roles in health and disease. Immunol. Rev. 2011, 242, 161–177.
- Wen, T.; Rothenberg, M.E. The Regulatory Function of Eosinophils. Microbiol. Spectr. 2016, 4.
- Ravin, K.A.; Loy, M. The Eosinophil in Infection. Clin. Rev. Allergy Immunol. 2016, 50, 214–227.
- Blanchard, C.; Rothenberg, M.E. Chapter 3 Biology of the Eosinophil. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2009; pp. 81–121.
- Kurose, N.; Mizuguchi, S.; Ohkanemasa, Y.; Yamashita, M.; Nakano, M.; Guo, X.; Aikawa, A.; Nakada, S.; Sasagawa, T.; Yamada, S. Adenosquamous carcinoma of the uterine cervix displaying tumor-associated tissue eosinophilia. SAGE Open Med. Case Rep. 2019, 7, 2050313X19828235.
- van Driel, W.J.; Hogendoorn, P.C.W.; Jansen, F.-W.; Zwinderman, A.H.; Trimbos, J.B.; Fleuren, G.J. Tumor-associated eosinophilic infiltrate of cervical cancer is indicative for a less effective immune response. Hum. Pathol. 1996, 27, 904–911.
- Höckel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schäffer, U.; Vaupel, P. Association between Tumor Hypoxia and Malignant Progression in Advanced Cancer of the Uterine Cervix1. Cancer Res. 1996, 56, 4509–4515.
- Xie, F.; Meng, Y.-H.; Liu, L.-B.; Chang, K.-K.; Li, H.; Li, M.-Q.; Li, D.-J. Cervical Carcinoma Cells Stimulate the Angiogenesis through TSLP Promoting Growth and Activation of Vascular Endothelial Cells. Am. J. Reprod. Immunol. 2013, 70, 69–79.
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607.
- Grisaru-Tal, S.; Rothenberg, M.E.; Munitz, A. Eosinophil–lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat. Immunol. 2022, 23, 1309–1316.
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426.
- Sakref, C.; Bendriss-Vermare, N.; Valladeau-Guilemond, J. Phenotypes and Functions of Human Dendritic Cell Subsets in the Tumor Microenvironment. Methods Mol. Biol. 2023, 2618, 17–35.
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory Molecules and Immune Checkpoints Are Differentially Expressed on Different Subsets of Dendritic Cells. Front. Immunol. 2019, 10, 1325.
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764.
- Kumar, V.; Bauer, C.; Stewart, J.H. Chasing Uterine Cancer with NK Cell-Based Immunotherapies. Future Pharmacol. 2022, 2, 642–659.
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2023, 23, 90–105.
- Tu, Y.; Pan, M.; Song, S.; Hua, J.; Liu, R.; Li, L. CD3+CD56+ natural killer T cell infiltration is increased in cervical cancer and negatively correlated with tumour progression. Biotechnol. Biotechnol. Equip. 2019, 33, 1380–1391.
- Céspedes, M.A.; Rodríguez, J.A.; Medina, M.; Bravo, M.; Cómbita, A.L. Analysis of NK Cells in Peripheral Blood and Tumor Infiltrating Lymphocytes in Cervical Cancer Patients. Rev. Colomb. Cancerol. 2012, 16, 16–26.
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661.
- Spaans, V.M.; Peters, A.A.W.; Fleuren, G.J.; Jordanova, E.S. HLA-E expression in cervical adenocarcinomas: Association with improved long-term survival. J. Transl. Med. 2012, 10, 184.
- Das, D.; Sarkar, B.; Mukhopadhyay, S.; Banerjee, C.; Mondal, S.B. An Altered Ratio of CD4+ And CD8+ T Lymphocytes in Cervical Cancer Tissues and Peripheral Blood—A Prognostic Clue? Asian Pac. J. Cancer Prev. 2018, 19, 471–478.
- Sheu, B.-C.; Hsu, S.-M.; Ho, H.-N.; Lin, R.-H.; Torng, P.-L.; Huang, S.-C. Reversed CD4/CD8 ratios of tumor-infiltrating lymphocytes are correlated with the progression of human cervical carcinoma. Cancer 1999, 86, 1537–1543.
- Jiang, W.; Kang, L.; Lu, H.-Z.; Pan, X.; Lin, Q.; Pan, Q.; Xue, Y.; Weng, X.; Tang, Y.-W. Normal Values for CD4 and CD8 Lymphocyte Subsets in Healthy Chinese Adults from Shanghai. Clin. Vaccine Immunol. 2004, 11, 811–813.
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4+FOXP3+ regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell. Mol. Immunol. 2011, 8, 59–66.
- Zhang, J.; Meng, S.; Zhang, X.; Shao, K.; Lin, C. Infiltration Patterns of Cervical Epithelial Microenvironment Cells During Carcinogenesis. Front. Immunol. 2022, 13, 888176.
- Lin, W.; Niu, Z.; Zhang, H.; Kong, Y.; Wang, Z.; Yang, X.; Yuan, F. Imbalance of Th1/Th2 and Th17/Treg during the development of uterine cervical cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 3604–3612.
- Nishimura, T.; Nakui, M.; Sato, M.; Iwakabe, K.; Kitamura, H.; Sekimoto, M.; Ohta, A.; Koda, T.; Nishimura, S. The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother. Pharmacol. 2000, 46, S52–S61.
- van der Burg, S.H.; Piersma, S.J.; de Jong, A.; van der Hulst, J.M.; Kwappenberg, K.M.C.; van den Hende, M.; Welters, M.J.P.; Van Rood, J.J.; Fleuren, G.J.; Melief, C.J.M.; et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc. Natl. Acad. Sci. USA 2007, 104, 12087–12092.
- Maskey, N.; Thapa, N.; Maharjan, M.; Shrestha, G.; Maharjan, N.; Cai, H.; Liu, S. Infiltrating CD4 and CD8 lymphocytes in HPV infected uterine cervical milieu. Cancer Manag. Res. 2019, 11, 7647–7655.
- Sanif, R.; Nurwany, R. Prognostic significance of CD4/CD8 ratio in patients with advanced cervical cancer. J. Phys. Conf. Ser. 2019, 1246, 012053.
- Zhang, Y.; Yu, M.; Jing, Y.; Cheng, J.; Zhang, C.; Cheng, L.; Lu, H.; Cai, M.-C.; Wu, J.; Wang, W.; et al. Baseline immunity and impact of chemotherapy on immune microenvironment in cervical cancer. Br. J. Cancer 2021, 124, 414–424.
- Li, L.; Xu, X.T.; Wang, L.L.; Qin, S.B.; Zhou, J.Y. Expression and clinicopathological significance of Foxp3 and VISTA in cervical cancer. Am. J. Transl. Res. 2021, 13, 10428–10438.
- Wu, M.-Y.; Kuo, T.-Y.; Ho, H.-N. Tumor-infiltrating lymphocytes contain a higher proportion of FOXP3+ T lymphocytes in cervical cancer. J. Formos. Med. Assoc. 2011, 110, 580–586.
- Ni, H.; Zhang, H.; Li, L.; Huang, H.; Guo, H.; Zhang, L.; Li, C.; Xu, J.-X.; Nie, C.-P.; Li, K.; et al. T cell-intrinsic STING signaling promotes regulatory T cell induction and immunosuppression by upregulating FOXP3 transcription in cervical cancer. J. ImmunoTherapy Cancer 2022, 10, e005151.
- Nedergaard, B.S.; Ladekarl, M.; Thomsen, H.F.; Nyengaard, J.R.; Nielsen, K. Low density of CD3+, CD4+ and CD8+ cells is associated with increased risk of relapse in squamous cell cervical cancer. Br. J. Cancer 2007, 97, 1135–1138.
- Tang, Y.; Zhang, A.X.J.; Chen, G.; Wu, Y.; Gu, W. Prognostic and therapeutic TILs of cervical cancer—Current advances and future perspectives. Mol. Ther.-Oncolytics 2021, 22, 410–430.
- Hong, H.S.; Mbah, N.E.; Shan, M.; Loesel, K.; Lin, L.; Sajjakulnukit, P.; Correa, L.O.; Andren, A.; Lin, J.; Hayashi, A.; et al. OXPHOS promotes apoptotic resistance and cellular persistence in TH17 cells in the periphery and tumor microenvironment. Sci. Immunol. 2022, 7, eabm8182.
- Alves, J.J.P.; De Medeiros Fernandes, T.A.A.; De Araújo, J.M.G.; Cobucci, R.N.O.; Lanza, D.C.F.; Bezerra, F.L.; Andrade, V.S.; Fernandes, J.V. Th17 response in patients with cervical cancer. Oncol. Lett. 2018, 16, 6215–6227.
- Oomizu, S.; Arikawa, T.; Niki, T.; Kadowaki, T.; Ueno, M.; Nishi, N.; Yamauchi, A.; Hirashima, M. Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin. Immunol. 2012, 143, 51–58.
- Punt, S.; van Vliet, M.E.; Spaans, V.M.; de Kroon, C.D.; Fleuren, G.J.; Gorter, A.; Jordanova, E.S. FoxP3(+) and IL-17(+) cells are correlated with improved prognosis in cervical adenocarcinoma. Cancer Immunol. Immunother. 2015, 64, 745–753.
- Yu, Q.; Lou, X.-M.; He, Y. Preferential Recruitment of Th17 Cells to Cervical Cancer via CCR6-CCL20 Pathway. PLoS ONE 2015, 10, e0120855.
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241.
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory Effects of B Cells on Antitumor Immunity. Cancer Res. 2006, 66, 7741–7747.
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674.
- Kim, S.S.; Shen, S.; Miyauchi, S.; Sanders, P.D.; Franiak-Pietryga, I.; Mell, L.; Gutkind, J.S.; Cohen, E.E.W.; Califano, J.A.; Sharabi, A.B. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin. Cancer Res. 2020, 26, 3345–3359.
- Chen, Z.; Zhu, Y.; Du, R.; Pang, N.; Zhang, F.; Dong, D.; Ding, J.; Ding, Y. Role of Regulatory B Cells in the Progression of Cervical Cancer. Mediat. Inflamm. 2019, 2019, 6519427.
This entry is offline, you can click here to edit this entry!