Red blood cells (RBCs) have been implicated in the progression of a wide range of disease states where their roles have been specifically linked to their adhesiveness. Such diseases include, inter alia, atherosclerosis, tumors (in terms of its growth suppression), polycythemia vera, central retinal occlusion and diabetes mellitus,
- blood cell adhesiveness
- blood cell hemolysis
- blood cell ghosting
1. Introduction
2. The Role of the Red Blood Cell in the Progression of Atherosclerosis
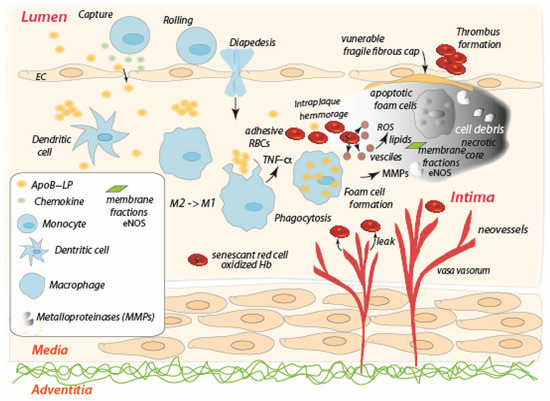
3. The Role of the Red Blood Cell in Tumor Necrosis
4. The Role of the Red Blood Cell in Other Diseases
5. Siderocytes and Macrophages
De Back et al. [83] describe the possible role of splenic macrophages in facilitating RBC vesiculation, a mechanism important for the removal of inclusions such as Heinz bodies (aggregates of denatured, oxidatively damaged hemoglobin) and regarding siderocytes, i.e., RBCs with one or more granular inclusion containing ferric Fe [84]. Macrophages have a pivotal role in this mechanism as they interact with aged, senescent, or diseased, RBCs that expose adhesion molecules, leading to vesiculation, lysis and ghost formation. For aged RBCs, i.e. siderocytes, the process is a self-protective mechanism that ends with vesiculation [85][86]. Adhesion strength is a fundamental factor to proceed along RBC vesiculation–lysis–ghosting pathway [1][87]. There is much to understand the details of this adhesion–hemolysis–ghost formation paradigm that influences RBCs life and the RBCs role(s) in a wide range of diseases.
6. Discussion
The paradigm of adhered RBCs subject to modest shear flows leading to vesiculation, hemolysis, and ghost formation, provides a unifying scenario of the RBCs’ road to clearance and of RBC role in disease progression and/or regression. It is possible to hypothesize that details of RBC adhesiveness may provide a diagnostic set of tools for assessing, and even designing, therapeutic strategies and methods. The combination of several technological approaches would provide more complete information useful to fully document the time scales of adhesiveness and its outcomes vis-a-vis hemolysis and ghost formation to understand RBCs adhesiveness in health and disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241210382
References
- Asaro, R.J.; Cabrales, P. Red blood cells: Tethering, vesiculation, and disease in micro-vascular flow. Diagnostics 2021, 11, 971.
- Pretini, V.; Koenen, M.H.; Kaestner, L.; Fens, M.H.A.M.; Schiffelers, R.M.; Bartels, M.; Van Wijk, R. Red Blood Cells: Chasing Interactions. Front. Physiol. 2019, 10, 945.
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282.
- Wautier, M.P.; Héron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055.
- Grossin, N.; Wautier, M.P.; Picot, J.; Stern, D.M.; Wautier, J.L. Differential effect of plasma or erythrocyte AGE-ligands of RAGE on expression of transcripts for receptor isoforms. Diabetes Metab. 2009, 35, 410–417.
- Kucukal, E.; Ilich, A.; Key, N.S.; Little, J.A.; Gurkan, U.A. Red Blood Cell Adhesion to Heme-Activated Endothelial Cells Reflects Clinical Phenotype in Sickle Cell Disease. Am. J. Hematol. 2018, 93, 1050–1060.
- Wautier, J.L.; Wautier, M.P. Cellular and Molecular Aspects of Blood Cell-Endothelium Interactions in Vascular Disorders. Int. J. Mol. Sci. 2020, 21, 5315–5330.
- Wautier, M.P.; El Nemer, W.; Gane, P.; Rain, J.D.; Cartron, J.P.; Colin, Y.; Le Van Kim, C.; Wautier, J.L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood 2007, 110, 894–901.
- El Nemer, W.; De Grandis, M.; Brusson, M. Abnormal adhesion of red blood cells in polycythemia vera: A prothrombotic effect? Thromb. Res. 2014, 133 (Suppl. 2), S107–S111.
- Alapan, Y.; Little, J.A.; Gurkan, U.A. Heterogeneous red blood cell adhesion and deformability in sickle cell disease. Sci. Rep. 2014, 4, 7173.
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells: A complex love story. Front. Physiol. 2014, 5, 9–17.
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561.
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355.
- Srinivasan, S.R.; Radhakrishnamurthy, B.; Vijayagopal, P.; Berenson, G.S. Proteoglycans, lipoproteins, and atherosclerosis. Adv. Exp. Med. Biol. 1991, 285, 373–381.
- Hurt-Camejo, E.; Camejo, G.; Rosengren, B.; López, F.; Ahlström, C.; Fager, G.; Bondjers, G. Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler. Thromb. 1992, 12, 569–583.
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138.
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46.
- Tabas, I.; Li, Y.; Brocia, R.W.; Xu, S.W.; Swenson, T.L.; Williams, K.J. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation. J. Biol. Chem. 1993, 268, 20419–20432.
- Kruth, H.S.; Jones, N.L.; Huang, W.; Zhao, B.; Ishii, I.; Chang, J.; Combs, C.A.; Malide, D.; Zhang, W.Y. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 2005, 280, 2352–2360.
- Tziakas, D.N.; Chalikias, G.; Pavlaki, M.; Kareli, D.; Gogiraju, R.; Hubert, A.; Böhm, E.; Stamoulis, P.; Drosos, I.; Kikas, P.; et al. Lysed Erythrocyte Membranes Promote Vascular Calcification. Circulation 2019, 139, 2032–2048.
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969, Erratum in: Nat. Rev. Immunol. 2010, 10, 460.
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325.
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232.
- Kamei, M.; Carman, C.V. New observations on the trafficking and diapedesis of monocytes. Curr. Opin. Hematol. 2010, 17, 43–52.
- Johnson, J.L.; Newby, A.C. Macrophage heterogeneity in atherosclerotic plaques. Curr. Opin. Lipidol. 2009, 20, 370–378.
- Paulson, K.E.; Zhu, S.N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 2010, 106, 383–390.
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55.
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediators Inflamm. 2012, 2012, 693083.
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349.
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108.
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852.
- de Vries, M.R.; Quax, P.H. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506.
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Role of lipids and intraplaque hypoxia in the formation of neovascularization in atherosclerosis. Ann. Med. 2017, 49, 661–677.
- Zemplenyi, T.; Crawford, D.W.; Cole, M.A. Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis 1989, 76, 173–179.
- Garcia-Touchard, A.; Henry, T.D.; Sangiorgi, G.; Spagnoli, L.G.; Mauriello, A.; Conover, C.; Schwartz, R.S. Extracellular proteases in atherosclerosis and restenosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1119–1127.
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061.
- Levy, A.P.; Moreno, P.R. Intraplaque hemorrhage. Curr. Mol. Med. 2006, 6, 479–488.
- Lang, F.; Abed, M.; Lang, E.; Föller, M. Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 2014, 21, 138–153.
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Föller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651.
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90.
- Setty, B.N.; Betal, S.G. Microvascular endothelial cells express a phosphatidylserine receptor: A functionally active receptor for phosphatidylserine-positive erythrocytes. Blood 2008, 111, 905–914.
- Comporti, M.; Signorini, C.; Buonocore, G.; Ciccoli, L. Iron release, oxidative stress and erythrocyte ageing. Free Radic. Biol. Med. 2002, 32, 568–576.
- Buttari, B.; Profumo, E.; Riganò, R. Crosstalk between red blood cells and the immune system and its impact on atherosclerosis. Biomed. Res. Int. 2015, 2015, 616834.
- Buttari, B.; Profumo, E.; Cuccu, B.; Straface, E.; Gambardella, L.; Malorni, W.; Genuini, I.; Capoano, R.; Salvati, B.; Riganò, R. Erythrocytes from patients with carotid atherosclerosis fail to control dendritic cell maturation. Int. J. Cardiol. 2012, 155, 484–486.
- Profumo, E.; Buttari, B.; Petrone, L.; Straface, E.; Gambardella, L.; Pietraforte, D.; Genuini, I.; Capoano, R.; Salvati, B.; Malorni, W.; et al. Redox imbalance of red blood cells impacts T lymphocyte homeostasis: Implication in carotid atherosclerosis. Thromb. Haemost. 2011, 106, 1117–1126.
- Williams, H.J.; Fisher, E.A.; Greaves, D.R. Macrophage differentiation and function in atherosclerosis: Opportunities for therapeutic intervention? J. Innate Immun. 2012, 4, 498–508.
- Maldonado, N.; Kelly-Arnold, A.; Vengrenyuk, Y.; Laudier, D.; Fallon, J.T.; Virmani, R.; Cardoso, L.; Weinbaum, S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: Potential implications for plaque rupture. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H619–H628.
- Kucukal, E.; Little, J.A.; Gurkan, U.A. Shear dependent red blood cell adhesion in microscale flow. Integr. Biol. 2018, 10, 194–206.
- Jani, V.P.; Asaro, R.; Oronsky, B.; Cabrales, P. RRx-001 Increases Erythrocyte Preferential Adhesion to the Tumor Vasculature. Int. J. Mol. Sci. 2021, 22, 4713–4724.
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–663.
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946–1959.
- Na, Y.R.; Je, S.; Seok, S.H. Metabolic features of macrophages in inflammatory diseases and cancer. Cancer Lett. 2018, 413, 46–58.
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46–69.
- van Dalen, F.J.; van Stevendaal, M.H.M.E.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9.
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell Longev. 2016, 2016, 2795090.
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914.
- Oršolić, N.; Kunštić, M.; Kukolj, M.; Gračan, R.; Nemrava, J. Oxidative stress, polarization of macrophages and tumour angiogenesis: Efficacy of caffeic acid. Chem. Biol. Interact. 2016, 256, 111–124.
- Virág, L.; Jaén, R.I.; Regdon, Z.; Boscá, L.; Prieto, P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox. Biol. 2019, 26, 101261.
- Regdon, Z.; Robaszkiewicz, A.; Kovács, K.; Rygielska, Ż.; Hegedűs, C.; Bodoor, K.; Szabó, É.; Virág, L. LPS protects macrophages from AIF-independent parthanatos by downregulation of PARP1 expression, induction of SOD2 expression, and a metabolic shift to aerobic glycolysis. Free Radic. Biol. Med. 2019, 131, 184–196.
- Dai, L.; Bhargava, P.; Stanya, K.J.; Alexander, R.K.; Liou, Y.H.; Jacobi, D.; Knudsen, N.H.; Hyde, A.; Gangl, M.R.; Liu, S.; et al. Macrophage alternative activation confers protection against lipotoxicity-induced cell death. Mol. Metab. 2017, 6, 1186–1197.
- Kim, B.C.; Kim, H.G.; Lee, S.A.; Lim, S.; Park, E.H.; Kim, S.J.; Lim, C.J. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem. Pharmacol. 2005, 70, 1398–1407.
- Kuo, P.L.; Chen, C.Y.; Hsu, Y.L. Isoobtusilactone A induces cell cycle arrest and apoptosis through reactive oxygen species/apoptosis signal-regulating kinase 1 signaling pathway in human breast cancer cells. Cancer Res. 2007, 67, 7406–7420.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282.
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296.
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139.
- Wu, S.; Li, T.; Liu, W.; Huang, Y. Ferroptosis and Cancer: Complex Relationship and Potential Application of Exosomes. Front. Cell Dev. Biol. 2021, 9, 733751.
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593.
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083.
- Sable, K.A.; Shields, B.E. The Role of Dietary Antioxidants in Melanoma and Nonmelanoma Skin Cancer. Cutis 2023, 111, 33–48.
- Remigante, A.; Spinelli, S.; Marino, A.; Pusch, M.; Morabito, R.; Dossena, S. Oxidative Stress and Immune Response in Melanoma: Ion Channels as Targets of Therapy. Int. J. Mol. Sci. 2023, 24, 887.
- Rogers, S.; McIntosh, R.L.; Cheung, N.; Lim, L.; Wang, J.J.; Mitchell, P.; Kowalski, J.W.; Nguyen, H.; Wong, T.Y.; International Eye Disease Consortium. The prevalence of retinal vein occlusion: Pooled data from population studies from the United States, Europe, Asia, and Australia. Ophthalmology 2010, 117, 313–319.
- Hirabayashi, K.; Tanaka, M.; Imai, A.; Toriyama, Y.; Iesato, Y.; Sakurai, T.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Kawate, H.; Tanaka, M.; et al. Development of a Novel Model of Central Retinal Vascular Occlusion and the Therapeutic Potential of the Adrenomedullin-Receptor Activity-Modifying Protein 2 System. Am. J. Pathol. 2019, 189, 449–466.
- Spivak, J.L. Polycythemia vera: Myths, mechanisms, and management. Blood 2002, 100, 4272–4290.
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665.
- Tan, X.; Shi, J.; Fu, Y.; Gao, C.; Yang, X.; Li, J.; Wang, W.; Hou, J.; Li, H.; Zhou, J. Role of erythrocytes and platelets in the hypercoagulable status in polycythemia vera through phosphatidylserine exposure and microparticle generation. Thromb. Haemost. 2013, 109, 1025–1032.
- Asaro, R.J.; Cabrales, P. The RBCs road to ghost and removal: Splenic clearance. Blood Adv. 2021, 5, 4422–4425.
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48.
- Kaur, C.; Foulds, W.S.; Ling, E.A. Blood-retinal barrier in hypoxic ischaemic conditions: Basic concepts, clinical features and management. Prog. Retin. Eye Res. 2008, 27, 622–647.
- Shin, E.S.; Sorenson, C.M.; Sheibani, N. Diabetes and retinal vascular dysfunction. J. Ophthalmic Vis. Res. 2014, 9, 362–373.
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J.; et al. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746.
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R.; Of macrophages and red blood cells: A complex love story.. Front Physiol 2014, 5, 9-17, .
- Crosby, W.H.; Siderocytes and the spleen.. Blood 1957, 12, 165-170, .
- Willekens, F.L.; Roerdinkholder-Stoelwinder, B.; Groenen-Döpp, Y.A.; Bos, H.J.; Bosman, G.J.; van den Bos, A.G.; Verkleij, A.J.; Were, J.M; Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation.. Blood 2003, 101, 747-751, .
- Willekens, F.L.; Were, J.M.; Groenen-Döpp, Y.A.; Roerdinkholder-Stoelwinder, B.; de Pauw, B.; Bosman, G.J.; Erythrocyte vesiculation: A self-protective mechanism?. Br J Haematol 2008, 141, 549–556, .
- Asaro, R.J.; Zhu, Q.; MacDonald, I.C.; Tethering, evagination, and vesiculation via cell-cell interactions in microvascular flow. . Biomech Model Mechanobiol 2020, 20, 31-53, .