Biofouling is the growth of organisms on wet surfaces. Biofouling includes micro- (bacteria and unicellular algae) and macrofouling (mussels, barnacles, tube worms, bryozoans, etc.) and is a major problem for industries. However, the settlement and growth of some biofouling species, like oysters and corals, can be desirable. Thus, it is important to understand the process of biofouling in detail. Modern “omic” techniques, such as metabolomics, metagenomics, transcriptomics, and proteomics, provide unique opportunities to study biofouling organisms and communities and investigate their metabolites and environmental interactions. Because "omics" originate from biomedical research and especially work at the cellular level, the learning curve for work in the environment is steep. We envision that as use of "omics" techniques especially combining different "omics" to address complex issues like biofouling will be transformational.
[1]1. Introduction
Pioneering studies of Prof. Claude E. ZoBell and his colleagues from the Scripps Institution of Oceanography (USA) provided the first information about the diversity of marine microbes in marine biofilms. Culture-dependent techniques were developed that allowed researchers to isolate and investigate the properties of marine bacteria for years. However, less than 1% of marine bacteria can be cultivated in the laboratory using traditional techniques [
50]. Thus, culture-dependent techniques limit our understanding of microbial diversity. Modern metagenomic techniques allow the identification of all microbes from biofilms and seawater and estimate their abundances (
Table 1).
The majority of metagenomic studies use amplicon sequencing of small pieces (300–1000 bp) of 16S ribosomal DNA for prokaryotes and pieces of 18S ribosomal DNA (300 plus bp) and pieces of COI (about 800 bp, cytochrome oxidase I) for eukaryote identification in biofouling communities. Metagenomics is the study of DNA sequences from microbes and metazoans [
51]. A key step is the use of the polymerase chain reaction (PCR) for making many copies of existing sequences of DNA that have been extracted from your environmental sample. For PCR, you use specific DNA “primer” sequences that you add that enable you to amplify specific genes [
52]. The amplified DNA is called amplicons. You use amplicons to identify organisms (microbes, animals, or plants) by comparison of the obtained DNA sequences with ones in the database. There are many databases but the most used ones are NCBI (National Institutes of Health, USA) and RDP (Ribosomal Database Project from Michigan State University, USA).
The first marine metagenomic study was conducted in 1991 by Dr. Norman R. Pace and colleagues who cloned 16S DNA of the picoplankton community from the Pacific Ocean [
53]. A few years later, one of the first metagenomics studies of biofilms was conducted on sulfate-reducing bacteria from marine sediments [
54]. In this study, 16S DNA genes were cloned in
E. coli and identified using Sanger sequencing. The development of sequencing techniques (
Table 1) and the reduction in costs resulted in many biofouling studies that utilize metagenomic approaches, which are reviewed using selected examples below.
Table 1. Summary of sequencing platforms used in metagenomic studies.
2. Metagenomics of Biofilms on Man-Made Substrata
Biofilms are a significant part of biofouling. Knowing the composition of the biofilm community is important for environmental toxicology, forensics, understanding surface microbe interaction, management, and ecology. For example, investigators using high throughput 454 pyrosequencing, based upon the detection of light released in the time of nucleotide incorporation during the polymerase chain reaction [
58] of 16S DNA genes. Analysis of pyrosequencing results demonstrated that bacterial communities developed on black and white panels exposed to fouling in the sea were different [
39]. However, classes Alphaproteobacteria and Firmicutes dominated in all biofilms. Another study that used the same next-generation sequencing (NGS) technique investigated the composition of microbial communities developed in a bioreactor [
59]. The investigators found that microbial diversity decreased under high aeration. Similarly, results of pyrosequencing demonstrate that aeration affected the diversity and species richness of bacterial and archaeal communities on osmosis membranes [
40].
Illumina is next-generation, high-throughput “sequencing by synthesis technology” based on tracking the addition of fluorescently labeled nucleotides during DNA polymerization [
58]. Illumina technology has frequently been used in biofouling metagenomic studies for the analysis of species composition of microbial biofilms developed on man-made structures [
60,
61,
62]. Analysis of microbial communities developed in membrane bioreactors via Illumina sequencing showed that high salinity increased the proportion of
Flavobacterium,
Aequorivita,
Gelidibacter,
Microbacterium, and
Algoriphagus genera [
41]. Another study investigated biofouling developed on sea gliders using 16S and 18S amplicon sequencing [
63]. The researchers observed differences in the number of OTUs (operational taxonomic units) between biofilms on different parts of the glider. Bacteria belonging to the classes gamma- and alpha-proteobacteria dominated prokaryotic communities, while hydrozoans and Chlorophyta dominated eukaryotic communities. Distinct bacterial communities were detected using MiSeq Illumina on stainless steel exposed to biofouling in a Northern Portugal port [
64]. In contrast, bacterial communities developed on stainless steel, polyethylene and titanium investigated using MiSeq Illumina shared some similarities [
65]. However, the communities changed over time. Most studies using NGS technology report that microbial communities developed on submerged surfaces differ from those in seawater [
63,
66].
High throughput sequencing using the Illumina platform is used in studies of metabolic assembled genomes from biofouling communities. For example, a study by Walter et al. [
67] of microbial mats of a coastal lagoon showed that they are dominated by cyanobacteria responsible for photosynthesis, Chroococcales responsible for nitrogen and ammonia assimilation, and Desulfobacterales contributing to sulfate reduction. Taxonomic and functional metagenomic analysis of biofilms developed in different locations and their effect on larval settlement of the polychaete,
Hydroides elegans, was investigated [
68]. The investigators demonstrated that the microbial communities were significantly different in coastal waters as compared to off-shore waters. However, the functional genes were similar between sites and related to carbohydrates, amino acids, and protein metabolism.
The temporal shift of microbial communities on wood and concrete used for artificial reefs was investigated using Ion Torrent sequencing technology [
69]. This technology is based on detecting hydrogen ions during DNA polymerization [
70]. The investigators found that the relative abundances of bacterial phyla decreased differently on different substrata over time [
69]. Similarly, microbial communities developed in brackish waters on different classes of stainless steel showed that one type of steel had different microbial communities dominated by Actinobacteria, while Proteobacteria dominated other types. Ion Torrent sequencing technology is also used to examine microorganisms on membranes [
42] and in bioreactors [
71].
MinION Oxford nanopore is a new sequencing technology (
Table 1). It can be used for amplicon sequencing as well as for the assembly of full genomes. The advantage of this technique is that it does not require a PCR step and can detect femtograms of DNA [
72]. It may be so that because this technology is novel, only a few biofouling-related studies were found (
Table 1). Complete genomic sequences of biofouling bacteria
Vibrio cambelli [
57,
73] and
Pseudomonas putida [
74] were determined using this technology. Similarly, the genome of important fouling species was sequenced using Illumina and Oxford nanopore technologies [
43].
NGS technology can be used to assess the composition of biofilms and investigate their effect on the settlement of macrofoulers. Biofilms and their mussel-inducing activity were investigated via Illumina MiSeq [
75]. The phylum Proteobacteria dominated all biofilms. The composition of biofilms inducing
Mytilus coruscus settlement was different from low inductive ones. The coral settlement-inducing activity of biofilms of crustose coralline alga was investigated using Illumina metagenomics and isolation of bacteria [
76]. Data analysis revealed no correlation between inductive settlement capacities and species of bacteria. A recent study demonstrated that biofilms formed in different environmental conditions affect the formation of macrofouling communities [
77]. Biofilms were developed in areas of high and low anthropogenic impact and then were translocated. A low settlement rate of non-indigenous species
Watersipora subatra on biofilms developed in marine protected areas and moved to the area with high anthropogenic impact. This demonstrates that metagenomics can be a useful tool in marine conservation.
Many studies investigated biofouling communities developed on plastics floating in the oceans or deposited in sediments using metagenomic approaches [
78,
79,
80]. In general, the studies confirmed the presence of diverse and specific microbial communities associated with different types of plastic which were different from microbes in the water column. Thus, microbial biofilms on the surface of plastic are called the “plastisphere” [
78]. Additionally, omic studies are used to identify species responsible for the degradation of plastics [
81].
3. Metagenomics of Biofilms on Antifouling Coatings and Biocides
Metagenomics of biofilms on antifouling coatings opened up a new world for researchers and industries. First, the studies indicated that microbial communities contain many species of prokaryotic and eukaryotic organisms. Diverse microbial communities were observed during a 1-year study of microbial biofilms on 11 biocidal antifouling coatings in Oman waters [
61]. Communities were dominated by alpha- and gamma-Proteobacteria, Cyanobacteria, and Flavobacteria. Similarly, clear differences between communities representing three different types of coatings were observed. The 454 pyrosequencing of 16S rRNA demonstrated spatiotemporal changes in microbial communities on different antifouling coatings in French waters [
82].
Second, metagenomic studies enable the identification of biofouling species present on fouling management coatings. The 454 pyrosequencing of 16S rRNA was used to evaluate microbes in biofilms on different coatings in French coastal waters [
82]. The study reports that the biocidal coating reduced the abundance of Bacteroidetes. Similarly, clear differences in microbial communities developed on ZnO nanorod coatings and copper-based coating on fishing nets were recorded using Illumina MiSeq technology in Oman coastal waters [
83]. The 16S and 18S amplicon Illumina MiSeq sequencing were used to evaluate prokaryotic and eukaryotic communities on different substrata in New Zealand waters [
84]. The authors found that the orientation of the substrate, and the presence of antifouling coatings, impacted the composition of microbial communities. Similarly, clear differences in prokaryotic and eukaryotic communities on antifouling coatings were detected in another study [
62]. However, in a different study, only prokaryotic communities were different between toxic and non-toxic coatings on sea gliders [
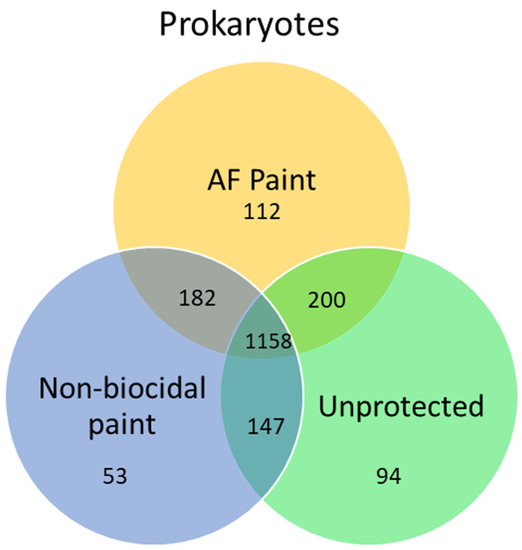
63]. This study reported that the majority of bacteria species (1158) were shared between different protected and un-protected surfaces (
Figure 1).
Figure 1 is a Venn diagram, which is a graphical representation of microbial community analysis. Additionally, principal component analysis and clustering algorithms can be employed. The highest number of unique operational taxonomic units (OTUs) were observed on biocidal antifouling paint, while the lowest OTUs were found in non-biocidal paint. In another study, marine biofilms on biocidal and non-biocidal antifouling coatings were studied using 16S amplicon MiSeq sequencing [
60]. Genera
Loktanella,
Sphingorhabdus, and
Erythrobacter dominated biocidal coatings, while
Portibacter dominated the fouling release ones. For decades people working with roof shingle fouling focused on one cyanobacterium
Gloeocapsa spp., and when dust samples from shingles were analyzed
Gloeocapsa proved to be a minor biofouling component [
35]. Shingle manufacturers interested in understanding their biofouling problem could take advantage of microbiome analysis and use it to develop an effective antifouling defense.
Figure 1. Venn diagram showing the number of shared and unique OTUs in prokaryotic communities studied via MiSeq Illumina and developed on sea gliders during the experiment of Dobretsov et al. [
63] AF is a copper-based antifouling paint. Non-biocidal paint is a chitosan-based paint. Unprotected are not painted parts of the glider.
Metagenomics is a powerful tool to detect the toxicity of coatings because of differences between microbial communities developed on toxic and non-toxic substrates. This can be used in forensic studies to detect compounds that are not listed in the recipes for industrial products. Ward et al. [
85] reported the results of microbial growth on seven different kinds of plastic preproduction pellets in seawater. The investigators report that biofilms on plastic pellets were initially different and remained different on pellets, especially on PVC (polyvinil chloride) which was different from all the rest of the microbiomes on pellets at every sample interval. After 70 days, there were four distinct microbiomes on the pellets, with the convergence of microbiomes on similar plastics. The authors found that groups of bacteria associated with toxic fouling management coatings were found on some plastics at all time intervals suggesting compounds leaching from the plastic had a role in biofilm community composition. The more we understand microbial communities using metagenomics the better we will be able to manage human health, ecosystem health, and food security and develop effective antifouling and antimicrobial defenses.
4. Environmental DNA (e-DNA)
One use of metagenomics for metazoans in the biofouling community is environmental DNA (e-DNA) [
86]. The idea is that DNA in water from near a fouling community can be filtered and then one can selectively amplify particular genes, such as Cytochrome Oxidase I (COI) or 18S ribosomal DNA, to determine members of the fouling community including invasive and cryptic species [
87]. Obtained sequences can be compared with publicly available data and groups of interest can be identified. One advantage of this approach is that one does not need to be an expert in systematics because there are extensive public databases containing systematic information that can be accessed and used.
Several groups worldwide have developed this approach to make lists of the species in their biofouling communities. For example, e-DNA was used to detect the invasive golden mussel
Limnoperna fortunei in Chinese waters [
87]. Another study used e-DNA from plastic litter to detect four invasive species [
88]. However, there are challenges associated with this technique. The e-DNA technique is too new to answer questions about the presence or absence of particular species and estimate their abundance at the time of sampling. No one knows the e-DNA shedding rates for different kinds of organisms. The sequences of some biofouling species are either absent in the databases or primers (for COI and 18S) and this prevents distinguishing them from other similar species. The DNA distribution in water is patchy [
89]. The methods of sampling and DNA extraction can have an impact on the results [
90]. Finally, all e-DNA are not equal and in complex communities, some dominant species can camouflage the presence of rare species.
There are a lot of important unknowns in the development and use of “omic” techniques in the environment. For example, the half-life of arginine carboxy-terminal signal peptides in marine environments is approximately 2 h. The half-life of e-DNA is in a similar range. However, though e-DNA might still be present, the pieces could be too short to be useful to be amplified as intact sequences of DNA hundreds of base pairs in length are required to identify organisms. More generally, the question of shedding rates and forms of shedding for different kinds of metazoans remains open. For example, do mollusks produce the same amount of e-DNA per gram of living animal as arthropods or polychaetes, or cnidaria? There is plenty to do to improve “omic” techniques in the future.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241310518