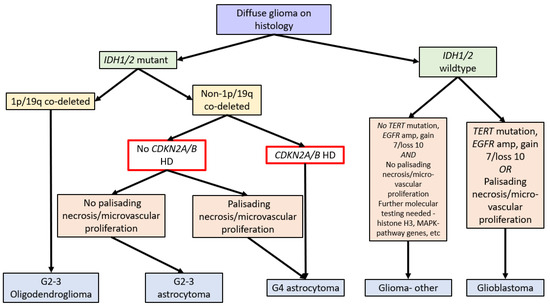
In 2020, the Consortium to Inform Molecular and Practical Approaches to CNS Tumour Taxonomy (cIMPACT-NOW), upgrade 5, published recommendations for grading criteria and terminologies in
-mutant astrocytomas. After reviewing the literature on multiple potential prognostic biomarkers, including
amplification, reduced global DNA methylation, genomic instability (high copy number variants or somatic mutations), and mitotic activity and proliferation indices, they concluded that while “significant mitotic activity” should remain as a criterion for distinguishing grade 3 from grade 2
HD, necrosis, or microvascular proliferation was present, a grade 4 designation was appropriate [
].
A prime example of supporting literature is Reis et al who were one of the earliest to report on the prognostic impact of
CDKN2A HD in the setting of
IDH mutations
. They identified
CDKN2A deletions as a prognostic marker specifically in
IDH-mutant grade 2 and 3 gliomas. The authors analysed 270 gliomas and identified
CDKN2A deletions via FISH in 57/108 grade 2 astrocytomas, 31/61 grade 3 astrocytomas, 23/96 oligodendrogliomas, and 19/49 oligoastrocytomas, inclusive of both homozygous and heterozygous
CDKN2A deletions. The authors assessed tumours for 1p/19q deletion if they were not morphologic astrocytomas and assessed all tumours for
IDH1/2 mutations by genome sequencing. They reported worse overall survival in grade 2 and 3 gliomas after adjusting for age, sex, and
IDH mutation (HR 1.6, 95% CI = 1.0–2.4,
p = 0.03). This significance was maintained in the astrocytoma subgroup (HR 2.0, 95% CI 1.1–3.5,
p = 0.02) but not for oligodendrogliomas or oligoastrocytomas (HR 0.7, 95% CI 0.2–2.0,
p = 0.5 and HR 0.8, 95% CI 0.3–2.4,
p = 0.7, respectively). Again, a portion of these morphologic oligodendrogliomas in this cohort would no longer be classified as such without the corresponding molecularly confirmed 1p19q co-deletion. Interestingly, the presence of deletions in the
IDH-mutant/
ATRX expression loss astrocytoma group, without
TP53 mutation, was non-prognostic (
p = 0.2) [
68]. Furthermore, as
ATRX loss and
TP53 mutations are strongly associated with
IDH-mutant astrocytomas, it is unclear what this
ATRX/
TP53 discordance represents in
IDH-mutant gliomas. Interestingly, given the FISH probe used covers a broad genomic region at 9p21,
CDKN2B status can be said to be assessed by proxy.
4.3.3. Literature That Counters CDKN2A/B Stratification
Not all studies supported the use of
CDKN2A/B in
IDH-mutant astrocytomas. One such example is Roy et al. who analysed the 9p region lost in malignancies by analysing two cohorts (the first group being 10,985 samples from 33 different cancer types and the second group being 540 low-grade gliomas from three databases) and reported that
CDKN2A inactivation did not promote tumour aggressiveness. Even when accounting for
IDH and 1p/19q status (
IDH-mutant 1p/19q non-deleted astrocytoma), there was no survival impact of CKDN2A HD. While they did show that heterozygous loss was associated with poor OS, mRNA expression was not altered. It was therefore postulated that this survival impact was due to the loss of other 9p genes [
30]. It is unclear why this report differs from the majority of other studies, but it highlights that not all studies support the role of
CDKN2A/B HD as a prognostic marker in
IDH-mutant astrocytomas.
5. Management of Tumours with CDKN2A/B Homozygous Deletions
There is no clear consensus on the treatment of
IDH-mutant astrocytomas with
CDKN2A/B HD, and reports related to their management are scarce. Reflecting this ambiguity, the current joint American Society of Clinical Oncology and Society of Neuro-Oncology guidelines recommend grade 4 astrocytomas be treated with concurrent temozolomide-radiotherapy with sequential temozolomide or radiotherapy alone with sequential temozolomide [
70].
However, given the evidence that
CDKN2A/B HD alters tumour biology (increased angiogenesis and cell growth), it cannot assume that these tumours will be as susceptible to temozolomide as their non-deleted counterparts. Unfortunately, the evidence for treatment specifically for
CDKN2A/B HD astrocytomas is minimal. In 2000, Iwadate et al. investigated the relationship between
CDKN2A deletion, p16 expression, and chemosensitivity to 30 different cytotoxic agents in vitro. They analysed 56 astrocytoma specimens (based on morphologic criteria,
IDH status unknown) and found 17 specimens had p16 alterations (
CDKN2A HD = 7,
CDKN2A mutation = 5, p16 loss on IHC = 5). When looking at samples with p16 alterations, they found that deletions correlated with increased sensitivity to anti-metabolite agents but not to alkylating agents, antibiotics, topoisomerase inhibitors, or anti-microtubule agents [
71].
6. Conclusions
CDKN2A/B HD have a direct oncogenic effect through loss of cell cycle inhibition and other parallel processes and are a molecular marker that influences grading and survival in IDH-mutant astrocytomas. Overall, the evidence supports the use of CDKN2A/B HD as a negative prognostic marker in IDH-mutant astrocytomas. However, there is a significant variation in certainty, methods used for deletion detection, and the quality of the presented literature. There are also inaccuracies resulting from misclassification of tumours in older studies based on the revised WHO classification. These limitations hamper conclusions regarding the certainty and depth of impact CDKN2A/B HD has on prognosis and management and how this impact is affected by other co-occurring molecular alterations. Therefore, the strongest evidence for CDKN2A/B HD in IDH-mutant astrocytomas must come from prospective reports with the current WHO 2021 classification.