Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Allergy
Asthma is a common respiratory disease that affects people of all ages, characterized by considerable heterogeneity in age, clinical presentation, genetics, epigenetics, environmental factors, treatment response, and prognostic outcomes. Asthma affects more than 330 million people worldwide, of which 33% are children under 14 years, and 27% are adults whose first symptoms occurred in childhood. However, the genetic and epigenetic mechanisms of childhood allergic diseases and asthma are still not fully understood.
- childhood asthma
- genetic factors
- epigenetics
1. DNA Methylation and Studies of DNA Methylation and Childhood Asthma
The best-described mechanism in the literature is CpG-DNA methylation [85]. DNA methyltransferases added a methyl group to cytosine in a CpG dinucleotide, converting cytosine to 5’-methylcytosine. Over 80% of CpG dinucleotide sites are located outside of CpG islands [93]. Methylation of CpG islands near the promoters leads to transcription repression, while hypomethylation leads to genomic instability, upregulation of gene expression, and elevated mutation rates [94].
The associations between DNA methylation and diseases can be identified by an epigenome-wide association study (EWAS). In EWAS, where thousands of CpGs are being evaluated, multiple testing issues should be addressed to reduce false-positive findings [95].
EWAS are primarily cross-sectional studies and cannot differentiate whether epigenetic changes precede the disease or are a consequence of it. According to available data on childhood asthma, blood-based EWAS provides information mainly at the eosinophilic level. However, nasal epithelium EWAS provides information on methylation changes and regulatory perturbations of epithelial cells in the respiratory tract [96]. In addition, nasal epithelium EWAS was more reproducible in other cohorts than EWAS conducted in blood samples. All these data suggest that the methylation is tissue- and cell-specific [97]. Therefore, several EWAS studies and projects have attempted to describe the DNA methylation of different cell types and tissues of asthma patients [85,94,96,98].
A significant overlap of methylation changes was found in nasal and bronchial epithelial cells of children with asthma [96]. When conducting blood-based EWAS and nasal epithelium EWAS, genes such as ACOT7, EPX, GJA4, and METTL were found to be common [99]. This indicates that, except for the cell specificity of CpG methylation sites, there is also an epigenetic effect across tissues [100].
Recent studies by Xu [101] and Qi [102] focused on epigenetics and childhood asthma development in detail. The studies described gene/genetic variants associated with childhood asthma and epigenetic modifications connected with asthma development. All data discussed above [100,101,103,104,105,106,107,108,109,110,111] are systematized in Table 3 (modified from Qi et al., 2019).
Table 3. Summary of the major childhood asthma DNA methylation studies.
| Gene/Candidate-Gene * | Ancestry | Tissue | Ref. |
|---|---|---|---|
| LMAN2; STX3; LPIN1; DICER1; SLC25A25; | European | Whole blood | Xu et al., 2018 [101] |
| ACOT7, EPX, GJA4 and METTL1; | American African | Nasal brush epithelial cells | Yang et al., 2018 [100] |
| ZFPM1; AP2A2; IL5RA; | European | Peripheral blood | Arathimos et al., 2017 [103] |
| CDHR3; CDH26; FBXL7; | Hispanic/Latino | Nasal brushes cells | Forno et al., 2018 [104] |
| EVL; NTRK1; SLC9A3; ACOT7; | Mixed ancestry | Nasal swab cells | Cardenas et al., 2019 [105] |
| CLNS1A, Mir_548; SUB1, LOC100129858; RUNX1; GPATCH2; WDR20; IL5RA; ACOT7; KCNH2; | -13 European cohorts; -3 mixed ancestry cohorts -1 African/American cohort |
Whole blood | Reese et al., 2019 [106] |
| ORMDL3; CCL26 | African American and European American | Bronchial airway epithelial cells | Nicodemus-Johnson et al., 2016 [107] |
| SMAD3; DDO/METTL24 | European | Whole blood | Lund et al., 2018 [108] |
| CCL5, IL2RA, TBX21, FCER2, TGFB1, LIF, ADAM17, NHEJ1, AHR, PGDR, PI3K, GNA12 etc. | American cohort | Peripheral blood | Rastogi et al., 2013 [109] |
| ARG1, ARG2, IL6, iNOS | non-Hispanic white and Hispanic white ethnicity | Buccal cells/Nasal epithelium | Breton et al., 2011 [110] |
| KRT5, CRIP1, STAT5A | European | Bronchial epithelium | Stefanowicz et al., 2012 [111] |
* Some genes were identified in more than one study.
2. Histone Modification and Studies of Histone Modification and Asthma
DNA condensation occurs with the help of core histones (H2A, H2B, H3, and H4) to form a chromatin structure [112]. Histone modifications are methylation, acetylation, phosphorylation, ubiquitination, sumoylation, and ADP-ribosylation. Disturbance in histone modifications has many clinical implications, leading to inflammatory responses by the immune system and the development of various diseases, including allergic asthma, rhinitis, atopic dermatitis, and allergies (Figure 2).
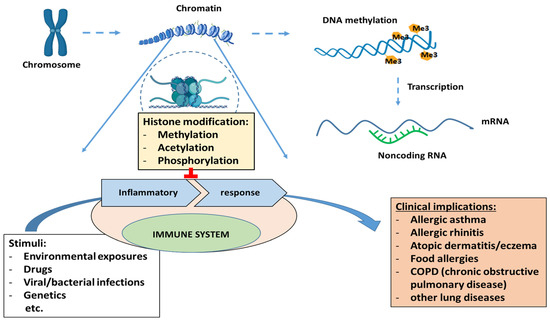
Figure 2. Clinical implications of epigenetic regulation (histone modifications, DNA methylation and non-coding RNAs).
Histone modifications alter chromatin accessibility. For example, histone acetylation is carried out by the enzyme histone acetyltransferase (HAT), leading to a loose chromatin structure. This makes it available to transcription factors and activates gene expression. On the other hand, histone deacetylation is carried out by the enzyme histone deacetylase (HDAC), resulting in gene silencing and a lack of gene expression. These histone modifications are associated with the risk of asthma development or disease severity (in children and adults). They affect the gene expression of genes responsible for the differentiation and maturation of various immune cells, some of which are involved in asthma development [113].
HAT activity has been increased in adults [114] and children with asthma [115], and the ratio of HAT/HDAC changes also depending on the presence or/and severity of asthma.
To date, no publications discuss in detail the role of histone modifications in the development of asthma in children. Some studies have been conducted in animal models [116,117,118,119]. The results of other studies established the role of HDACs in T-cell development and asthma susceptibility. Their suppressed expression can lead to asthma and/or allergic airway diseases with varying severity, as well as resistance to corticosteroid therapy [114,120]. In HDAC1-deficient T-cell mice, there is an increased number of eosinophils and the production of Th2 cytokines, which is a prerequisite for asthma development [121]. These findings indicate that HDACs are essential in repairing airway epithelium, especially in airway remodeling observed in severe asthma [122].
A genome-wide histone modification study from 12 asthma patients and 12 controls identified 200 enhancer regions in three immune cell subpopulations (naive T cells, Th1 and Th2 cells). In addition, a selective enrichment in demethylation of the Lys at the fourth position of the N-terminal tail of histone 3 (H3K4me2) was shown. This marker indicates gene activation in promoters and enhancers of genes [123]. One hundred sixty-three regions were specific for the Th2 cells, 29 were specific for naïve T cells, and only 11 for Th1 cells.
A study of children with allergic asthma (n = 14) and controls (n = 18) identified a high level of histone acetylation (histone 3 at the IL13 and FOXP3 loci). The study also indicates a potential regulatory role of IL13 acetylation on the amount of protein product [124].
Similarly, acetylation of histone H3 lysine 18 (H3K18) is related to increased expression of STAT6, EGFR, and ΔNp63 [125]. The expression of epidermal growth factor receptor (EGFR) is vital for the proliferation, differentiation, and repair processes. It was increased in both healthy and damaged areas of the epithelium of pediatric and adult patients with asthma [125,126,127,128]. In addition, the signal transducer and activator of transcription 6 (STAT6) are also overexpressed in the epithelium of patients with severe asthma [129,130].
In a recent study, loss of HAT was found to be associated with a downregulation of the ORMDL3 gene, which in turn led to the development of childhood asthma [131].
Some other studies have also identified a link between childhood asthma and histone modifications. For example, Wawrzyniak et al. found higher expression of histone deacetylases 1 and 9 in epithelial cells from 18 asthmatic patients and 9 controls [132]. In addition, increased expression of HDACs led to increased expression of IL-13 and IL-4, which are critical genes for childhood asthma severity and treatment response.
It is well-known that prenatal and/or childhood exposure to environmental factors can influence the immune system and responses [133,134]. In addition, environmental factors can strongly induce epigenetic changes in immune cells, leading to the development of allergies/asthma [135,136].
In 2011, Brand et al. investigated epigenetic alteration in pregnant mice and offspring [137]. They demonstrated that changing H4 acetylation led to a childhood asthma phenotype.
Another research group evaluated the acetylation of histones H3 and H4 in the promoter regions of six immunoregulatory genes associated with childhood allergy in 173 placentas [138]. Three genes were found to be associated with high histone acetylation levels and risk of allergic sensitization—H3 acetylation in placentas at the IFNG and SH2B3 genes and H4 acetylation at HDAC4. These data suggest that placental histone acetylation levels are associated with a reduced risk of allergen sensitization and have the potential to predict the development of childhood asthma/allergies.
Inhibition of HDAC activity has a preventive effect on epithelial barrier integrity in pediatric and adult patients with asthma [132].
To sum up, there is no doubt that histone modifications play a crucial role in the development and pathogenesis of asthma. However, more studies with larger cohorts are needed to obtain more accurate and transparent information about their contribution to immune-mediated and allergic diseases, such as asthma. Moreover, maternal asthma remains among the highest risk factors for childhood-onset asthma. Data show that epigenetic alterations may occur in the embryo of pregnant asthmatic women that may lead to severe asthma development in the offspring.
A summary of the studies on histone modification and childhood asthma [124,125,131,132,138] are presented in Table 4.
Table 4. Summary of the studies about the role of histone modification and childhood asthma.
| Genes/Candidate-Genes * | Histone Modifications | Tissue | Ref. |
|---|---|---|---|
| IL-13, FOXP3 | histone H3 acetylation | Peripheral blood | Harb et al., 2015 [124] |
| IL-4, IL-13 | higher expression of HDACs 1 and 9 | Bronchial epithelium | Wawrzyniak et al., 2016 [132] |
| STAT6, EGFR and ΔNp63 | histone H3 acetylation | Bronchial airway epithelial cells | Stefanowics et al., 2015 [125] |
| IFNG, SH2B3 HDAC4 |
histone H3 acetylation histone H4 acetylation |
Blood samples Placenta samples |
Harb et al., 2019 [138] |
| ORMDL3 | histone H3 acetylation | Bronchial epithelium | Cheng et al., 2019 [131] |
* Some genes were identified in more than one study.
3. Non-Coding RNAs and Studies of Non-Coding RNA and Asthma
Non-coding RNAs, such as microRNAs (miRNAs, miRs), are single-strand RNAs of 21–25 nucleotides that regulate gene expression at the post-transcriptional level [139]. They act as dynamic regulators of gene networks by exercising control over the translation of many mRNAs by mRNA destabilization [140]. miRNAs play a significant role in biochemical pathways, cell signaling, cell and tissue development, and regulating pathological processes in various diseases. They have been identified as related to childhood/adult asthma, expressed in different tissues, including blood cells, airway epithelial cells, and smooth muscle. They can also serve as potential biomarkers for diagnosis and therapy targets for asthma [141]. Differences in miRNA expression between asthmatic and healthy subjects were found [98]. Some asthma-related miRNAs may act on genes associated with respiratory tract function (i.e., miR-10, miR-140-3p, miR-708), and others may target genes related to immune system function (i.e., miR-21, miR-19a, miR-155, miR-210, miR-125b, miR-223) [142].
MiRNAs, such as miRNA-17-18-19-20, miRNA-26a/b, miRNA-27a/b, miRNA-125b, miRNA-155, and others, polarize macrophages as a reaction to specific environmental stimuli and signals toward an M1-phenotype (classically activated). Others as miRNA-21, miRNA-124, miRNA-223-3p, and miRNA-511-3p, e.g., polarize macrophages towards an M2-phenotype (alternatively activated) [143].
In a recent study, two miRNAs (miR-21 and miR-126) were found to be upstream-regulated in patients with asthma compared to controls [144]. Another study showed that miR-21 contributes to the production of eosinophils and proliferation [145]. MiRNA-21 is one of the most well-studied miRNAs in relation to asthma. In asthmatic patients, its high amount suppresses IL-12p35 and causes resistance to steroids [146]. IL-12p35 is predominantly produced by dendritic cells, monocytes, and macrophages and inhibits cytokine-induced STAT pathways and cell-cycle regulators. Serum miRNA-21 is related to asthma onset or allergies and could be used as a biomarker [146,147]. Both miRNA-21 and miRNA-146a were associated with increased levels of eosinophils and could be biomarkers for the eosinophilic endotype of asthma [148].
Another study identified increased levels of miRNA-155 and decreased levels of Let-7a in the blood of children with asthma (n = 100) compared to controls (n = 100) [149]. To distinguish the severity of the disease, different expression profiles of miRNA-155 and Let-7a were found [149]. MiRNA-155 plays a critical role in IL-33 regulation of airway inflammation in mouse models of asthma [150]. In addition, the Let-7 miRNA has been shown to target IL13 and exert anti-inflammatory effects in mice [151]. These data suggest that miRNA-155 could be one of the biomarkers suitable for childhood asthma diagnosis.
Another miRNA, miRNA-146, has also been studied in relation to asthma [152]. HLA-G variation in children also increased the risk of developing asthma, along with miRNA-152 [108]. In an examination of serum samples from 160 children in the Childhood Asthma Management Program (CAMP), 155 circulating miRNAs were detected [153]. Of these, eight were significantly related to PC20 (asthma test to determine the concentration of inhaled methacholine), but the most substantial relationship was with miRNA-296-5p. Two others, miRNA-30d-5p, and miRNA-16-5p, were found to be critical in the development of asthma via regulating airway smooth muscles [153].
MiRNAs may also be used as biomarkers to assess the risk of childhood asthma development or to predict the outcomes from treatment, as several studies showed [142,154,155,156]. All these findings demonstrated the importance of miRNAs in controlling inflammation, diagnosing disease severity, and treatment response in childhood asthma, as well as their potential use as disease biomarkers.
The miRNA studies associated with asthma [157,158,159,160,161,162,163,164] are summarized in Table 5.
Table 5. Summary of some significant miRNAs’ studies associated with asthma.
| miRNAs | Level | Function/Role in Asthma | Tissue | Ref. |
|---|---|---|---|---|
| miR-21 | increased | - eosinophilic accumulation - correlation with IL-13 - Th2 response - suppression of IL-12p35 |
bronchial epithelial cells | Lu et al., 2011 [157] Wu et al., 2014 [144] Pua et al., 2015 [145] Elbehidy R., 2016 [146] |
| miR-146 | increased | - increase the eosinophilic level - anti-inflammatory - negative regulation of IL1β and COX2 |
blood samples, human ASM cells |
Su et al., 2011 [152] Comer et al., 2014 [158] Hammad et al., 2018 [148] |
| miR-155 | increased | - decreased level of Let-7a - regulation of IL-13 - in Th2 immune responses |
blood samples | Zhou et al., 2016 [159] Karam et al., 2019 [149] |
| miR-19 | increased | -mediate the production of IL-13 -targets mRNA of the TGFβR2 gene -regulation in responses of the Th2 cell |
bronchial epithelial cells, airway T cells |
Simpson et al., 2014 [160] Haj-Salem et al., 20015 [161] |
| miR-126 | Increased | - increase AHR and immune migration - Th2 response - increase the eosinophilic level |
bronchial epithelial cells, peripheral blood |
Mattes et al., 2009 [162] Wu et al., 2014 [144] Tian et al., 2018 [163] |
| miR-15b, 126, -139, -142, -186 etc. |
- lung function parameters in children | Blood samples | Kho et al., 2016 [164] |
This entry is adapted from the peer-reviewed paper 10.3390/allergies3020009
This entry is offline, you can click here to edit this entry!