Heterocyclic compounds, which are made up of both carbon and non-carbon atoms, serve as a crucial structural foundation for numerous chemicals with pharmacological and biological value. The research on heterocyclic compounds is an important part of organic chemistry and is utilized extensively in many industries, especially medicine. Heterocyclic compounds serve as the main active ingredient in a variety of pharmaceuticals, including analgesics, anti-inflammatory drugs, anti-tubercular drugs, antihypertensives, antidepressants, and even anticancer drugs. Many novel heterocyclic targeted drugs have emerged.Molecular targeted therapy is a key element of the new era of comprehensive multidisciplinary cancer treatment. A considerable number of molecular targeted medications have been created as part of the development of targeted therapy.
- heterocyclic compounds
- molecular targeted drugs
- nanomedicine
- cancer
1. Drugs Acting on Cell Membranes
2. Drugs Acting on the Cytoplasm
3. Drugs Acting on the Cell Nucleus
| The Spatial Location of Target | Compound Name | Chemical Structure Formula | Targets [IC50 (μM)] | Cancer | Cell [IC/GI/CC50 (μM)] | Reference |
|---|---|---|---|---|---|---|
| Cell membrane | 1 | 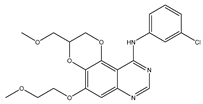 |
EGFR [IC50 = 10.29 × 10−3] |
NSCLC | A549 [IC50 = 9.95] |
[2] |
| NCI-H157 [IC50 = 11.66] |
||||||
| T293 [CC50 > 100] |
||||||
| WI-38 [CC50 = 90.55] |
||||||
| 2 | 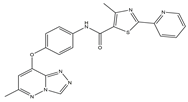 |
c-Met [IC50 = 90.00 × 10−3] |
Lung, liver and breast cancers | A549 [IC50 = 1.06 ± 0.16] |
[7] | |
| MCF-7 [IC50 = 1.23 ± 0.18] |
||||||
| HeLa [IC50 = 2.73 ± 0.33] |
||||||
| LO2 [IC50 > 50.00] |
||||||
| Cytoplasm | 3 |  |
HSP90 [IC50 = 110.18 × 10−3] |
Lung cancer | A549 [GI50 = 0.07 ± 0.01] |
[9] |
| H1975 [GI50 = 0.05 ± 0.01] |
||||||
| Hep3B [GI50 = 0.20 ± 0.03] |
||||||
| MDA-MB-231 [GI50 = 0.09 ± 0.01] |
||||||
| 4 | 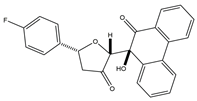 |
AKT [NA] |
Colon cancer | HCT-116 [IC50 = 0.92 ± 0.05] |
[12] | |
| Cell nucleus | 5 | 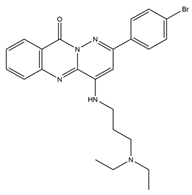 |
Topo I [NA] |
Ovarian, nasopharyngeal, stomach and lung cancers | SK-OV-3 [IC50 = 1.93 ± 0.16] |
[13] |
| CNE-2 [IC50 = 2.33 ± 0.52] |
||||||
| MGC-803 [IC50 = 1.39 ± 0.14] |
||||||
| NCI-H460 [IC50 = 1.55 ± 0.14] |
||||||
| LO-2 [IC50 = 5.71 ± 0.60] |
||||||
| 6 | 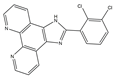 |
c-Myc G-quadruplex [NA] |
Nasopharyngeal cancer | CEN-1 [IC50 = 1.1 ± 0.1] |
[16] | |
| HaCaT [IC50 = 16.8 ± 0.7] |
||||||
| TME | 7 | 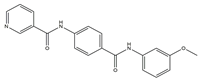 |
VEGFR-2 [IC50 = 2.17] |
Breast, liver and colon cancers | MCF-7 [IC50 = 1.37] |
[17] |
| HepG-2 [IC50 = 1.05] |
||||||
| HCT-116 [IC50 = 1.46] |
||||||
| WI-38 [IC50 = 60.8] |
||||||
| 8 | 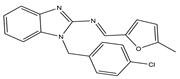 |
VEGFR-2 [NA] |
Lung metastasis of melanoma | NA | [18] | |
| Multiple targets | 9 | 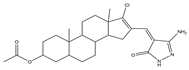 |
JAK2/Tubulin [NA] |
Lung cancer | A549 [IC50 = 27.36] |
[19] |
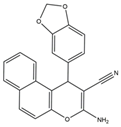
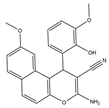
| 10–12 | 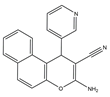 |
Topo I/Topo II/EGFR/VEGFR-2 [IC50 = 0.1392–0.6349] |
Breast, liver and colon cancers | MCF-7 [IC50 = 1.6–2.2] |
[20] | |
 |
HLF-1 [IC50 = 19.1–24.3] |
|||||
 |
WI-38 [IC50 = 20.2–26.5] |
| Compound | Name |
|---|---|
| 1 | N-(3-chlorophenyl)-5-(2-methoxyethoxy)-3-(methoxy-methyl)- 2,3-dihydro-[1,4]dioxino[2,3-f]quinazolin-10-amine |
| 2 | 4-Methyl-N-(4-((6-methyl-[1,2,4]triazolo[4,3-b]- pyridazin-8-yl)oxy)phenyl)-2-(pyridin-2-yl)thiazole-5-carboxamide |
| 3 | N-Ethyl-2,4-dihydroxy-5-isopropyl-N-(pyridin-3-yl)benzamide |
| 4 | (2R,5R)-5-(4-Fluorophenyl)-2-((S)-9-hydroxy-10-oxo-9,10-dihydrophenanthren-9- yl)dihydrofuran-3(2H)-one |
| 5 | 2-(4-Bromophenyl)-4-((3-(diethylamino)propyl)amino)-10Hpyridazino [6,1-b] quinazolin-10-one |
| 6 | 2-(2,3-dichlorophenyl)-1H-imidazo[4,5-f][1,10]phenanthroline |
| 7 | N-(4-((3-Methoxyphenyl)carbamoyl)phenyl)nicotinamide |
| 8 | 1-(4-chlorobenzyl)-2-(5-methyl-2-furfurylidenenamino)-benzimidazole |
| 9 | (Z)-16-((3-amino-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)methyl)-17-chloro-10,13-dimethyl-2,3,4,5,6,7,8,9,10,11,12,13,14,15-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl acetate |
| 10 | 3-Amino-1-(pyridin-3-yl)-1H-benzo[f]chromene-2-carbonitrile |
| 11 | 3-Amino-1-(benzo[d][1,3]dioxol-5-yl)-1H-benzo[f]chromene-2- carbonitrile |
| 12 | 3-Amino-9-methoxy-1-(2-hydroxy-3-methoxyphenyl)-1H-benzo [f]-chromene-2-carbonitrile |
4. Drugs Acting on the Tumor Microenvironment (TME)
5. Drugs Acting on Multiple Targets
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15061706
References
- London, M.; Gallo, E. Epidermal growth factor receptor (EGFR) involvement in epithelial-derived cancers and its current antibody-based immunotherapies. Cell Biol. Int. 2020, 44, 1267–1282.
- Qin, X.; Yang, L.; Liu, P.; Yang, L.; Chen, L.; Hu, L.; Jiang, M. Design, synthesis and biological evaluation of 2,3-dihydro- dioxino quinazoline derivatives as EGFR inhibitors. Bioorg. Chem. 2021, 110, 104743.
- Zhang, Q.; Zheng, P.; Zhu, W. Research progress of small molecule VEGFR/c-Met inhibitors as anticancer agents (2016-Present). Molecules 2020, 25, 2666.
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651.
- Wang, Z.; Dai, Z.; Wang, B.; Gao, Y.; Gao, X.; Wang, L.; Zhou, S.; Yang, L.; Qiu, X.; Liu, Z. Targeting c-MET to enhance the efficacy of Olaparib in prostate cancer. Onco Targets Ther. 2021, 14, 4383–4389.
- Wang, X.; He, L.; Wang, Y.; Liu, Y.; Wang, Y.; Chen, D.; Gong, D.; Fan, Y.; Wu, Y.S. MiR-300 alleviates cell proliferation and migration and facilitates cell apoptosis by targeting c-Met in gastric cancer. J. Oncol. 2022, 2022, 6167554.
- Zhang, Q.; Liu, X.; Gan, W.; Wu, J.; Zhou, H.; Yang, Z.; Zhang, Y.; Liao, M.; Yuan, P.; Xu, S.; et al. Discovery of triazolo-pyridazine/-pyrimidine derivatives bearing aromatic (heterocycle)-coupled azole units as class II c-Met inhibitors. ACS Omega 2020, 5, 16482–16490.
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in cancer. Int. J. Mol. Sci. 2021, 22, 10317.
- Liu, Y.M.; Tu, H.J.; Wu, C.H.; Lai, M.J.; Yu, S.C.; Chao, M.W.; Wu, Y.W.; Teng, C.M.; Pan, S.L.; Liou, J.P. Ring-opening of five-membered heterocycles conjugated 4-isopropylresorcinol scaffold-based benzamides as HSP90 inhibitors suppressing tumor growth in vitro and in vivo. Eur. J. Med. Chem. 2021, 219, 113428.
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88.
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54.
- Huang, J.; Chen, Y.; Guo, Y.; Bao, M.; Hong, K.; Zhang, Y.; Hu, W.; Lei, J.; Liu, Y.; Xu, X. Synthesis of dihydrofuran-3-one and 9,10-phenanthrenequinone hybrid molecules and biological evaluation against colon cancer cells as selective Akt kinase inhibitors. Mol. Divers. 2022, 27, 845–855.
- Huang, W.Y.; Zhang, X.R.; Lyu, L.; Wang, S.Q.; Zhang, X.T. Pyridazino quinazolinones as new anticancer scaffold: Synthesis, DNA intercalation, topoisomerase I inhibition and antitumor evaluation in vitro and in vivo. Bioorg. Chem. 2020, 99, 103814.
- Revikumar, A.; Kashyap, V.; Palollathil, A.; Aravind, A.; Raguraman, R.; Kumar, K.M.K.; Vijayakumar, M.; Prasad, T.S.K.; Raju, R. Multiple G-quadruplex binding ligand induced transcriptomic map of cancer cell lines. J. Cell. Commun. Signal. 2022, 16, 129–135.
- Lejault, P.; Moruno-Manchon, J.F.; Vemu, S.M.; Honarpisheh, P.; Zhu, L.; Kim, N.; Urayama, A.; Monchaud, D.; McCullough, L.D.; Tsvetkov, A.S. Regulation of autophagy by DNA G-quadruplexes. Autophagy 2020, 16, 2252–2259.
- Wu, Q.; Song, Y.; Liu, R.; Wang, R.; Mei, W.; Chen, W.; Yang, H.; Wang, X. Synthesis, docking studies and antitumor activity of phenanthroimidazole derivatives as promising c-myc G-quadruplex DNA stabilizers. Bioorg. Chem. 2020, 102, 104074.
- Ran, F.; Li, W.; Qin, Y.; Yu, T.; Liu, Z.; Zhou, M.; Liu, C.; Qiao, T.; Li, X.; Yousef, R.G.; et al. Inhibition of vascular smooth muscle and cancer cell proliferation by new VEGFR inhibitors and their immunomodulator effect: Design, synthesis, and biological evaluation. Oxid. Med. Cell. Longev. 2021, 2021, 8321400.
- Hsu, M.J.; Chen, H.K.; Chen, C.Y.; Lien, J.C.; Gao, J.Y.; Huang, Y.H.; Hsu, J.B.; Lee, G.A.; Huang, S.W. Anti-Angiogenetic and Anti-Lymphangiogenic Effects of a Novel 2-Aminobenzimidazole Derivative, MFB. Front. Oncol. 2022, 12, 862326.
- Tantawy, M.A.; Shaheen, S.; Kattan, S.W.; Alelwani, W.; Barnawi, I.O.; Elmgeed, G.A.; Nafie, M.S. Cytotoxicity, in silico predictions and molecular studies for androstane heterocycle compounds revealed potential antitumor agent against lung cancer cells. J. Biomol. Struct. Dyn. 2022, 40, 4352–4365.
- El-Mawgoud, H.K.A.; Fouda, A.M.; El-Nassag, M.A.A.; Elhenawy, A.A.; Alshahrani, M.Y.; El-Agrody, A.M. Discovery of novel rigid analogs of 2-naphthol with potent anticancer activity through multi-target topoisomerase I & II and tyrosine kinase receptor EGFR & VEGFR-2 inhibition mechanism. Chem. Biol. Interact. 2022, 355, 109838.