1. Drugs Acting on Cell Membranes
EGFR is widely distributed on cell surfaces and participates in modulating cell growth, proliferation, and differentiation [
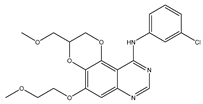
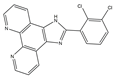
38]. A set of heterocyclic 2,3-dihydro-[1,4]dioxino [2,3-f] quinazoline derivatives to target EGFR were conceived and created by Qin et al. IC50 is the concentration of a drug that inhibits 50 percent of targets. All substances were shown to have the ability to inhibit EGFR kinase according to the enzyme assay results (IC
50 = 10.29–652.3 nM). Compared with erlotinib and gefitinib, compound
1 showed the strongest inhibition of EGFR kinase, with an IC
50 value of 10.29 nM. Anti-proliferation showed that compound
1 could inhibit the proliferation of A549 and NCI-H157 cell lines. The median cytotoxic concentration (CC50) is the concentration of a drug that causes 50% of normal cells to die. The larger difference between CC50 and IC50 values of tumor cells or between the IC50 values of normal and tumor cells, the safer and less toxic the drug. The CC50 of compound
1 against the human renal epithelial cell line T293 was larger than 100 μM, and the CC50 against the normal lung cell line WI-38 was 90.55 μM, indicating that compound
1 had no significant cytotoxic effect in vitro. Further studies demonstrated that compound
1 was joined to the ATP-binding pocket of the EGFR active site [
39].
It has been reported that c-Met targeting can be used in the treatment of NSCLC, prostate cancer, gastric cancer, and other tumors [
40,
41,
42,
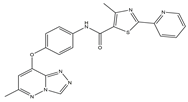
43]. New triazolo-pyridazine/pyrimidine derivatives were synthesized, evaluated, and found to inhibit c-Met kinase in A549, MCF-7, and HeLa cell lines that overexpressed c-Met. Among them, compound
2, the most effective compound, displayed obvious cytotoxicity to A549, MCF-7, and HeLa cell lines, with IC
50 values of 1.06 ± 0.16, 1.23 ± 0.18, and 2.73 ± 0.33 μM, respectively. Meanwhile, compound
2 significantly inhibited c-Met kinase activity, with an IC
50 value of 0.090 μM, similar to Foretinib. Acridine orange staining and flow cytometry analysis revealed that compound
2 could not only boost apoptosis in A549 cells but also trigger G0/G1 phase cell cycle arrest. Mechanically, the 5-methyl thiazole fragment is introduced into the five-atom fraction, which is essential for c-Met inhibition. Compound
2 could be a prospective class II c-Met inhibitor [
44].
2. Drugs Acting on the Cytoplasm
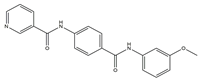
A protein-folding chaperone known as the heat shock protein (HSP) maintains the stability of numerous signal transduction proteins in cells to assist in cell growth and survival [
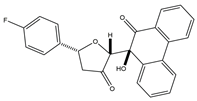
45]. HSP90 inhibitors, a group of ring-opening dihydroxy benzamide compounds, were devised and synthesized by Liu et al. Compound
3 showed the obvious inhibition of HSP90, with an IC
50 value of 110.18 nM as well as BIIB021, and led to the degradation of AKT downstream signaling, which was concentration- and time-dependent. The GI
50 values of compound
3 were 0.07 μM against the KRAS mutant A549 cell line and 0.05 μM against the EGFR mutant H1975 cell line. Compound
3 had stronger inhibition of cell proliferation and metastasis than 17-AAG and BIIB021. Compound
3 prevented A549 cells from migrating, with an IC
50 value of 1 μM. Compound
3 is a highly permeable drug with little toxic potency or cardiotoxicity. Pharmacokinetic tests showed that its oral bioavailability was 17.8%. Additionally, compound
3 exhibited anti-tumor activity at a daily dose of 50 mg/kg, with 72% tumor growth delay in a nude mouse A549 lung xenograft model. In the lung H1975 xenograft model, the combination therapy of compound
3 and afatinib resulted in a 67.5% tumor growth inhibition. Therefore, compound
3 is a promising potential lung cancer treatment medication [
46].
The dysfunction of the PI3K-AKT-mTOR signaling pathway can not only lead to neurodegeneration, cancer, and other diseases but also induce drug resistance [
47,
48]. Huang and colleagues created a series of dihydrofuran-3-one and 9, 10-phenquinone hybrid compounds. CCK-8 assay showed that these compounds had inhibitory effects on HCT-116, A549, and SJSA-1 cells, and the IC
50 value of HCT-116 cells was between 0.92 and 3.91 μM. Compound
4 was the most cytotoxic to HCT-116 cells and could arrest the cell cycle in the G2/M phase. Molecular docking assays revealed that compound
4 had a more significant inhibitory effect on AKT kinase than CAL-101. Compound
4 exerts its anti-tumor effects as an AKT kinase inhibitor by competitive binding to ATP to limit intradomain and interdomain motility. This result was further confirmed by a kinase selectivity assay [
49].
3. Drugs Acting on the Cell Nucleus
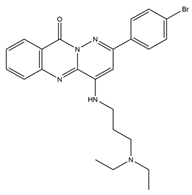
DNA topoisomerase (Topo), an enzyme in the nucleus, is involved in DNA replication, recombination, transcription, etc. Huang et al. designed and synthesized more than 30 pyridazino[1,6-b]quinazolinones derivatives. Compared with CPT and doxorubicin positive controls, compound
5 caused clear moderate or even potent cytotoxicity against the SK-OV-3, CNE-2, MGC-803, NCI-H460, and LO-2 cell lines (IC50 = 1.93 ± 0.16, 2.33 ± 0.52, 1.39 ± 0.14, 1.55 ± 0.14, 5.71 ± 0.60, respectively). Further studies revealed that most compounds could be embedded in DNA molecules. The Topo I inhibition assay indicated that Topo I was severely inhibited by compound
5. In a dose-dependent manner, compound
5 elicited mild G2 cell cycle arrest and apoptosis. In the MGC-803 xenograft tumor model, compound
5 had a significant inhibitory effect, with total growth inhibition reaching 55.9%. Additionally, molecular docking studies demonstrated that compound
5 might bind to Topo I and reduce its activity compared to doxorubicin. In conclusion, pyridazino[1,6-b]quinazolinones are novel drug scaffolds that can be used to produce anticancer medications [
50].
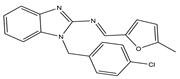
G-quadruplex (G4s) plays a significant role in biological activities by regulating telomerase function, gene expression, and the composition of topologically associated domains [
51,
52]. Using microwave-assisted technology, Wu et al. designed and synthesized a range of phenomidazole derivatives containing phenomidazole and imidazole heterocyclic aromatic rings, which are new drugs with anti-tumor activity. In comparison to doxorubicin, compound
6 inhibited nasopharyngeal carcinoma CNE-1 cell proliferation in a dose-dependent manner, with an IC
50 value of 1.1 μM. Compound
6 showed low toxicity to human keratinocyte HaCaT cells, with an IC
50 value of 16.8 μM. It could also induce apoptosis and G0/G1 cycle arrest in CNE-1 cells. Mechanistically, compound
6 could exert anti-tumor effects by stabilizing c-Myc G4s DNA, leading to DNA damage and thereby activating reactive oxygen species-mediated mitochondrial dysfunction. In the zebrafish xenograft model, compound
6 significantly inhibited the growth of xenograft tumors and had only a low toxic effect on zebrafish embryo development under the precondition of an effective concentration [
53].
4. Drugs Acting on the Tumor Microenvironment (TME)
The surrounding milieu of tumor cells is called the tumor microenvironment. It is made up of the extracellular matrix, bone marrow-derived inflammatory cells, fibroblasts, immune cells, and adjacent blood vessels. Using sorafenib as the lead compound, Ran et al. designed and synthesized three series of novel compounds containing pyridine and evaluated these compounds for VEGFR-2 suppression and anticancer activity with Sorafenib as the positive control. Compared with the control group, compound
7 exhibited significant cytotoxicity effects on MCF-7, HepG-2, HCT-116, and WI-38 cell lines (IC50 of 1.37, 1.05, 1.46, and 60.8 μM, respectively).. More importantly, compound
7 had the strongest inhibitory effect on VEGFR-2 (IC
50 = 2.17 μM). In contrast to the untreated group, further research revealed that compound
7 considerably up-regulated the expressions of caspase 3 and Bax while drastically downregulating the expression of Bcl-2 in HepG-2 cells. Compound
7 might encourage apoptosis and cause G2/M cell cycle arrest in HepG-2 cells according to flow cytometry studies. Additionally, when compared to celecoxib (87%), compound
7 considerably reduced the level of tumor necrosis factor α expression. At the same time, it also restrained the proliferation of vascular smooth muscle cells in a concentration-dependent manner. The binding of the compound to the ATP pocket of VEGFR-2 was found by the molecular docking method, which provided a basis for further elucidation of the structure–activity relationship [
54].
Similarly, compound
8 (MFB), a small molecule compound containing two amino benzimidazoles designed and synthesized by Hsu et al., might target VEGF/VEGFR to prevent angiogenesis and lymphangiogenesis. By blocking VEGF-A and VEGF-C signaling, compound
8 prevented human umbilical vein endothelial cells and lymphatic endothelial cells from proliferating, migrating, invading, and forming endothelial tubes in vitro. The results were also confirmed in animal experiments. Additionally, a mouse model of lung metastasis of melanoma confirmed that compound
8 showed an anti-tumor metastasis effect in vivo. Molecular docking experiments revealed that binding to VEGFR-2 might be the mechanism of compound
8 in inhibiting angiogenesis and lymphangiogenesis [
55].
5. Drugs Acting on Multiple Targets
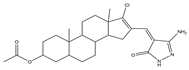
The importance of the JAK2/STAT3 signaling pathway in numerous disorders has been established by researchers. With the use of the A549 cell line as the experimental model, six 3′-acetoxy-5′-androsterane heterocyclic compounds were chosen for testing their antiproliferative effects on NSCLC. Compound
9 was the most toxic to A549 cells (IC
50 = 27.36 μM). According to the findings of molecular docking studies, the anticancer activity of compound
9 might be caused by its strong affinity for JAK2 and tubulin-colchicine soblidotin. The tubulin assay further verified the activity of compound
9 in inhibiting tubulin polymerization. According to flow cytometry research, compound
9 could not only accelerate the apoptosis of A549 cells but also halt the progression of the cell cycle, causing pre-G1 and G2/M cell cycle arrest. Additionally, compound
9 induced DNA fragmentation, up-regulation of apoptotic genes, down-regulation of anti-apoptotic genes, and mitochondrial dysfunction. Therefore, compound
9 is a multitarget anticancer drug that has great application potential [
56].
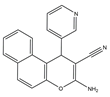
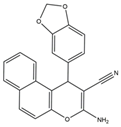
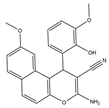
El-mawgoud et al. designed and synthesized novel rigid analogs of 2-naphtho and studied their biological activities. When tested on the breast cancer cell line MCF-7, colon cancer cell line HCT-116, and liver cancer cell line HepG-2, compounds
10–
12 showed inhibitory activity comparable to vinblastine and doxorubicin. Compounds
10 and
11 demonstrated the highest levels of cytotoxicity against MCF-7, HCT-116, and HepG-2 cell lines (IC
50 = 2.2, 2.4, and 2.4 μM, respectively, for compound
10, and 1.6, 1.5, and 5.5 μM, respectively, for compound
11). With an IC
50 value of 2.1 μM, compound
12 also demonstrated strong cytotoxicity against the breast cancer cell line MCF-7. In comparison to the control group, treatment groups with compounds
10,
11, or
12 increased MCF-7 cell apoptosis and G2/M cell cycle arrest. Mechanistically, they exerted strong anticancer effects by targeting Topo I and II, EGFR, and VEGFR-2 (IC50 = 0.1392–0.6349 μM). Compounds
10–
12 showed weak inhibitory activity against HFL-1 and WI-38 cells with IC50 values ranging from 19.1 to 26.5 μM. They were also characterized by good oral bioavailability and high capacity for transport. Together, these compounds have great potential for cancer therapy [
57].
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15061706