Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Ferroptosis, characterized by glutamate overload, glutathione depletion, and cysteine/cystine deprivation during iron- and oxidative-damage-dependent cell death, is a particular mode of regulated cell death. It is expected to effectively treat cancer through its tumor-suppressor function, as mitochondria are the intracellular energy factory and a binding site of reactive oxygen species production, closely related to ferroptosis.
- mitochondria
- ferroptosis
- cancer
- iron
- oxidative damage
1. Introduction
Nowadays, cancer is still one of the deadliest diseases in the world. According to the current trend of significant cancer development, the cancer incidence rate worldwide will double by 2070 [1]. Finding new ways to treat cancer has become an urgent matter. In addition, widely used therapies such as radiotherapy, chemotherapy, gene therapy, and immunotherapy are gradually being commonly used for the clinical treatment of tumors. However, these treatment modalities have specific adverse reactions or are challenging to pinpoint precisely, and patients need to be treated multiple times [2].
Inducing tumor cell death is one of the effective pathways for tumor therapy. With the continuous deepening of cancer research, it has been found that various forms of cell death, such as apoptosis and ferroptosis, can induce the death of cancer cells. Conducting a profound study on the death mode of tumor cells will play an essential role in the pathological mechanism of tumor development and treatment. Among these, ferroptosis, a newly discovered iron-dependent method of regulated cell death (RCD), is associated with the occurrence and treatment response of various types of tumors, and the ferroptosis pathway associated with cancer also affects the growth and proliferation of cancer cells [4]. Conversely, ferroptotic injury can promote macrophage M2 polarization and trigger immunosuppression of tumor-associated inflammation, thereby favoring tumor growth [4].
2. Hallmarks of Ferroptosis
2.1. Morphological Hallmarks
Morphological features are cellular variation characteristics that can be directly observed by optical or electron microscopy and do not require the use of special stains for staining. Ferroptosis is one mode of RCD, but unlike other modes of RCD, ferroptosis does not show similar morphological changes as observed in apoptosis. Still, necrotic characteristics such as plasma membrane rupture and cell swelling are often observed where ferroptosis occurs [15]. Yagoda et al. and Dixon et al. found that ferroptosis mainly cause mitochondrial membrane density increase, mitochondrial volume decrease, mitochondrial crest disappearance, and other mitochondrial-related morphological changes [11,16].
2.2. Biochemical Hallmarks
2.2.1. Iron Accumulation
Due to ferroptosis being an iron-dependent RCD, its biochemical characteristics are related to iron-accumulation-mediated biochemical events [20]. Dixon et al. found that iron chelators inhibit the occurrence of ferroptosis in vitro or in vivo [11]. Hou et al. observed that cellular labile iron concentration increases during ferroptosis induction [21]. Iron intake mediated by lactotransferrin (LTF), the transferrin receptor (TFRC), and transferrin (TF) can regulate ferroptosis by regulating ferroptosis sensitivity, which identifies certain molecular regulators related to iron homeostasis. However, whether different iron metabolism regulators play the same role in ferroptosis remains uncertain.
2.2.2. Lipid Peroxidation
Lipid peroxidation is an important biochemical marker for ferroptosis. More particularly, polyunsaturated fatty acids (PUFAs) oxidation by ROS to produce lipid hydroperoxides (L-OOH) is the most vital characteristic of ferroptosis [22]. Researchers found that ROS play a cell-type-dependent role in initiating ferroptosis and are produced by the Fenton reaction, which is mitochondria- or nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)-mediated [11,23,24]. The key enzymes of lipid peroxidation to initiate ferroptosis are the arachidonate lipoxygenase (ALOX) family, which includes ALOX5, ALOX3, ALOX12, ALOX15B, and ALOX15 in human cells [25,26].
2.2.3. Inhibition of Antioxidant Systems
Because LPO is the primary biochemical feature in ferroptosis, early discoveries of ferroptosis inducers such as erastin and RSL3 are associated with inhibiting antioxidant systems [11,29]. At present, researchers have confirmed three antioxidant systems, including the GSH, coenzyme Q10 (CoQ10), and tetrahydrobiopterin (BH4) pathways [11,30,31,32,33]. These systems can work separately or together to inhibit ferroptosis mediated by oxidative damage. The GSH system is the main pathway to limit ferroptosis. Erastin can inhibit the upstream regulator system Xc− (an amino acid antiporter) or the downstream effector glutathione peroxidase 4 (GPX4) of GSH from inducing ferroptosis [34].
2.3. Protein Concentration Changes
2.3.1. GPX4
GPX4 uses GSH as a substrate to reduce L-OOH to lipid alcohols (L-OH), inhibiting erastin-induced ferroptosis. Yang et al. also found ferroptosis inducers such as erastin and RSL3, which inactivate GPX4 to inhibit tumor growth in xenograft mice [29]. However, GSH depletion and inactivation of GPX4 resulted in increased LPO, leading to ferroptosis [36].
2.3.2. P53
The tumor suppressor p53 (TP53), through transcription and translation, produces P53, which is a cancer suppressor protein. In fibroblasts and specific cancer cells such as the human breast cancer cell line (MCF7) and human osteosarcoma cell line (U2OS), p53 inhibits the expression of SCL7A11. SCL7A11 promotes GSH synthesis by mediating cystine uptake and glutamate release, protects cells from oxidative stress, and prevents lipid peroxidation-induced ferroptosis [37]. When the concentration of P53 increases, it inhibits the biochemical effect of SCL7A11, which promotes the occurrence of ferroptosis and causes the death of cancer cells, thus playing a tumor-suppressive role. In addition, excess SCLS7A11 can inhibit P53-induced ferroptosis [38].
2.3.3. ACSL4
ACSL4 is a member of the long-chain family of ACSL, which is involved in the synthesis regulation of arachidonic acid-CoA (AA-CoA) and adrenic acid-CoA (AdA-CoA). Doll et al. found that ACSL4 is essential in shaping the cellular lipidome and determining sensitivity resistance to ferroptosis [27]. It leads to lipid peroxidation by inhibiting GPX4. Loss of ACSL4 is accompanied by a decrease in AA- and AdA-containing PE species utilized as substrates to induce the ferroptosis cascade of events when GPX4 is inactive [27].
3. Mechanisms of Ferroptosis
3.1. Mitochondrial Mechanisms of Ferroptosis
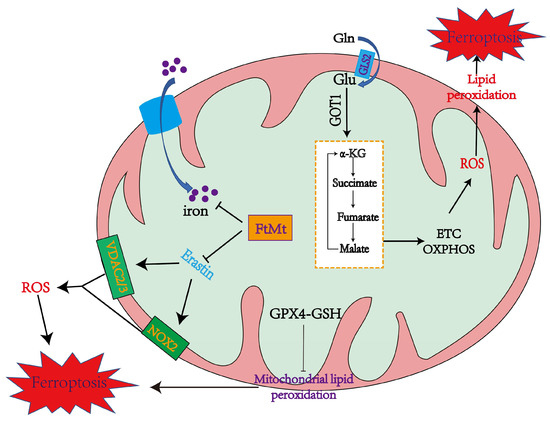
Cellular ROS are mainly produced by complexes I and III in the mitochondrial electron transfer chain (due to electronic leakage, complexes I and III have superoxide anion, which can promote lipid peroxidation and ferroptosis) [48,49]. 2-oxoacid dehydrogenase complexes (ODHc) can produce ROS at much higher rates than complex I, which has been shown in a number of papers [13]. It is composed of multiple copies of three enzyme components: oxoglutarate dehydrogenase (E1), dihydro-lipoamide succinyltransferase (E2), and dihydro-lipoamide dehydrogenase (E3) [13]. The ROS produced by ODHc during metabolism is mainly due to the reaction of its internal FAD/FADH2 (redox coenzyme) with oxygen molecules. FAD/FADH2 receive and release electrons during metabolism, which can combine with oxygen molecules to form highly reactive superoxide radicals (O2−), a type of ROS [50]. ROS production likely induces ferroptosis by accelerating the accumulation of lipid peroxidation. Mitochondrial GPX4 inhibits lipid peroxidation in mitochondria and diminishes ferroptosis (Figure 1).
Figure 1. The mechanisms of mitochondria in promoting and inhibiting ferroptosis. Mitochondria coordinate necroptotic cell death through multiple mechanisms. The mitochondrial glutaminolysis-TCA-ETC axis is a significant pathway to induce ferroptosis, during which the production of ROS leads to ferroptosis through lipid peroxidation. GPX4 also inhibits lipid peroxidation in mitochondria. FtMt stores iron and inhibits Erastin from upregulating NOX2, VDAC2, and VDAC3 to alleviate ferroptosis [14].
Mitochondrial ferritin (FtMt) can stockpile iron and is located in cellular mitochondria, and has been demonstrated to be structurally and functionally similar to the cytosolic H-ferritin [51]. The expression of FtMt is confined to cellular mitochondria of the central nervous system and some other tissues with high oxygen consumption [52]. Erastin upregulates VDACs located in the mitochondrial membrane, which induces the occurrence of ferroptosis [11]. However, there is no change in VDAC2 and VDAC3 levels in erastin-treated FtMt-SY5Y cells, indicating that FtMt protects erastin-induced cells from ferroptosis in a specific way [11,53]. NOX is upregulated in erastin-treated cells, which could be vital in generating ROS in ferroptosis [11].
3.2. GPX4 and Lipid Metabolism Mechanisms in Ferroptosis
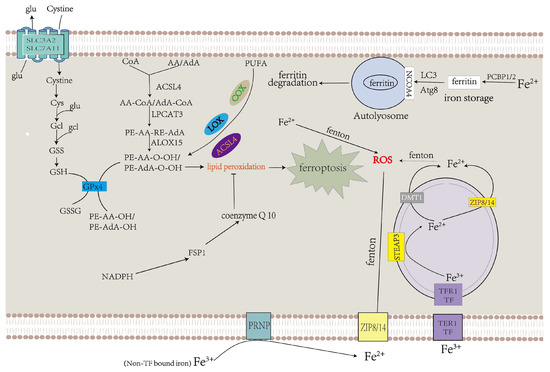
GPX4 could alleviate cellular ferroptosis by providing mitochondria with GSH, a reductant that promotes the elimination of cellular ROS [55]. Extracellular cysteine could be transported into the cell via system Xc− [11]. Cystine is converted to cysteine and binds with glutamate to form γ-glutamyl-cysteine (γGC) under glutamyl-cysteine ligase (GCL), a key enzyme in the process of GSH synthesis. After that, GSH synthase catalyzes the connection between γGC and glycine to form GSH [56]. GPX4 could convert GSH into GSH disulfide and transform noxious L-OOH into avirulent L-OH [57]. L-OOH also contributes to ferroptosis through another pathway. Free PUFAs and CoA are linked to form PUFA-CoAs under the action of ACSL4, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) catalyzes the binding of AA/AdA-CoA and membrane PE to produce AA/AdA–PE. ALOXs promote the conversion of AA/AdA–PE to PE-AA-O-OH/PE-AdA-O-OH, thus resulting in ferroptosis [58]. As a tumor suppressor protein, p53 represses the expression of SLC7A11, a crucial component of system Xc− (Figure 2).
Figure 2. Abnormalities in iron and lipid metabolism induce ferroptosis. Insufficiently controlled intracellular or degraded iron storage and PUFA-enriched phospholipids are the premises for ferroptosis leading to cell death. System Xc− transfers cystine into the cytoplasm, which is converted to cysteine and used to produce GSH, a necessary ingredient of GPX4 for eliminating LPO. CoQ10 also decelerates LPO [64].
Physiologically, PUFAs constantly undergo redox to achieve equilibrium. When oxidation processes surpass reduction processes, which exceed the cell’s capacity, this can result in plethoric lipid peroxidation and ferroptosis [11,59]. PUFAs can be oxidized under the function of multifarious lipoxygenases (LOX), cyclooxygenases (COX), and ACSL4, which promote lipid peroxidation (Figure 2).
3.3. Iron Metabolism in Ferroptosis
Under physiological circumstances, ferric ion (Fe3+) is transported into the cell under transferrin receptor 1 (TFR1), which takes up transferrin-bound iron (TBI) into cells by receptor-mediated endocytosis and then transforms into ferrous iron (Fe2+) via six-transmembrane epithelial antigen of the prostate 3 (STEAP3) in the endosome [65,66]. Compared with Fe3+, Fe2+ is more toxic to cells. Ferrous iron is transferred from the endosome to the cytoplasm via DMT1 [67]. ZIP8 and ZIP14 can transport non-transferrin-bound iron (NTBI) across the cell membrane, and this process is intensified by ferrireductase in the prion protein PRNP which reduces Fe3+ to Fe2+ as one of its multiple functions [68,69,70,71,72,73]. Superfluous Fe2+ results in the accumulation of ROS by the Fenton reaction, which facilitates the occurrence of ferroptosis (Figure 2) [74].
4. Mitochondrial Function in Ferroptosis
4.1. The Role of Mitochondria in Inducing Ferroptosis
4.1.1. Mitochondrial ROS Production and Lipid Peroxidation in Ferroptosis
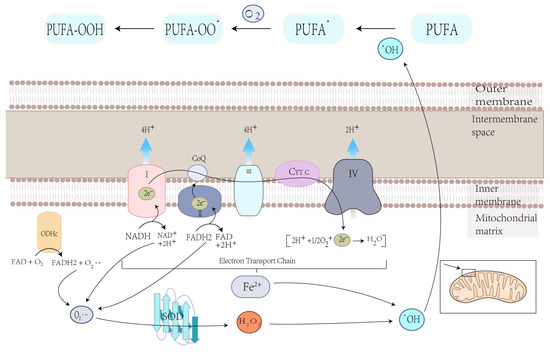
Mitochondria play a crucial role in the production of ROS, the specialized molecules that regulate cell signaling and function and performs several essential functions [48]. Carsten Culmsee et al. demonstrated that mitochondrial ROS are significantly increased in HT-22 and MEF cells when stimulated with erastin [82]. Ferroptosis is the result of lipid peroxidation of the cell membrane, which ensures the participation of ROS, and mitochondria play an essential role in this process [56]. O2•−, produced in mitochondria, is converted to hydroxyl radicals (•OH) through complex processes. This hydroxyl radical (•OH) later extracts bis-allylic hydrogen from PUFAs to form lipid radical which reacts with oxygen to form PUFA-OO• and ultimately PUFA hydroperoxides (PUFA-OOH) (Figure 3) [83,84]. At the same time, excess iron can damage cells due to ROS production, eventually leading to ferroptosis [85]. Therefore, the production of ROS in mitochondria may lead to lipid peroxidation on cell membranes and, ultimately, ferroptosis.
Figure 3. Electron leakage from the ETC cycle and the metabolic press of OGHDc produce O2•−, which is then converted to H2O2 under superoxide dismutase (SOD) mediation. H2O2 reacts with Fe2+ to produce hydroxyl radicals (•OH), which can extract the bis-allylic hydrogen in PUFAs from PUFA radicals (PUFA•). Then, PUFA• reacts with oxygen from PUFA peroxyl radicals (PUFAOO•), which extracts hydrogen from another lipid to generate PUFA hydroperoxides (PUFA-OOH).
4.1.2. The Mitochondrial TCA Cycle May Cause Ferroptosis
Glutamine can be used as a nitrogen source for the anabolism of various substances and a carbon source for TCA in mitochondria, and mitochondrial glutaminase GLS breaks down glutamine to produce glutamate. Glutamate is then converted by glutamic-oxaloacetic transaminase (GOT1) to α-KG, fueling the TCA recycle process [86]. Studies have shown that inhibiting glutaminolysis can lead to ferroptosis [23,87]. The TCA cycle may regulate ferroptosis by supporting electron transport chains (ETC) that promote ROS production. Another possible route is that ETCs drive proton motive force, and ATP synthesis which inhibits AMP-activated protein kinase (AMPK), the enzyme which can block ferroptosis by inhibiting the synthesis of malonyl-CoA (a precursor for PUFA synthesis) [88].
4.1.3. Iron Overload in Mitochondria Promotes Ferroptosis
Mitochondria are the centers of intracellular iron metabolism. The endosome-mitochondria “kiss and run” interaction is a significant pathway widely studied to transport iron into mitochondria [89,90]. Iron enters into the mitochondrial matrix through the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). Iron is stored in FtMt in the mitochondrial stroma or used to synthesize heme and Fe-S clusters. These two iron-containing factors are likely associated with ferroptosis [51]. The mechanism by which iron is transported through OMM is still unknown, while transport on the IMM requires the assistance of the membrane transporter mitoferrin 1 (Mfrn1) and its homolog mitoferrin 2 (Mfrn2) [91]. Friedreich’s ataxia (FRDA) is caused by a decrease in mitochondrial proteins frataxin (FXN), and one of the features is an iron overload on the mitochondria [92].
4.2. The Role of Mitochondria in Inhibiting Ferroptosis
There are three main types of cellular protective systems against ferroptosis. Each of these three protective systems has its own unique subcellular localization. GPX4 is localized to the cytoplasm and mitochondria, FSP1 is localized to the plasma membrane, and DHODH is localized to the mitochondria. Among them, the inhibition of ferroptosis by mitochondria is mainly through GPX4 and DHODH [96]. GPX4, a member of the glutathione peroxidase family, plays an important role in inhibiting ferroptosis. GPX4 is an inhibitor of lipid peroxidation, which can degrade small molecular peroxides and relatively complex lipid peroxides, and has shown inhibitory ability in adriamycin-induced lipid peroxidation [97].
5. Ferroptosis Inducer
5.1. Ferroptosis Inducers Targeting Different Mechanisms
5.1.1. Ferroptosis Inducers Targeting the Production of ROS via Iron Accumulation
Iron accumulation in mitochondria produces mitochondrial ROS via the Fenton reaction, which uses ferrous ions as catalysts to convert H2O2 in cells into ROS to induce ferroptosis [101]. DHA accelerates ferritin degradation and then promotes ROS accumulation in cells to induce ferroptosis by regulating the activity of the AMPK/mTOR/p70S6k signaling pathway [102]. Auriculasin, isolated from Flemingia philippinensis, induces ferroptosis of colorectal cancer cells by increasing the accumulation of Fe2+ which increases ROS production in cells [103]. In conclusion, targeting the production of ROS via iron accumulation to induce ferroptosis may be an effective cancer therapy.
5.1.2. Ferroptosis Inducers Targeting System Xc−
System Xc−, located in the cell membrane, mediates the transport of cysteine and glutamate. Glutamate is transferred to the outside of the cell, and simultaneously, cystine, which participates in the generation of GSH, is imported, thereby preventing the ferroptosis of cancer cells [104]. Therefore, some molecules inhibiting system Xc− via several targets have been indicated for treating cancers by inducing cancer cell ferroptosis. Erastin, specifically inhibiting the system, has been demonstrated to be effective in treating a variety of tumors, including meningioma, diffuse large B-cell lymphoma (DLBCL), and renal cell carcinoma [29,105,106]. With the exact mechanism of ferroptosis induced by erastin, sulfasalazine, approved by the FDA, has been widely used in the treatment of endometrial cancer [107,108].
5.1.3. Ferroptosis Inducers Targeting the Consumption of GSH
GSH is an essential member of the cellular antioxidative system, loss of which will break redox homeostasis and cause ROS accumulation, eventually triggering the ferroptosis of cancer cells [111]. Artesunate, used as an anti-malarial drug previously, has been repurposed as an anticancer drug due to its induction of cell death in head and neck cancer via inducing ferroptosis by decreasing cellular GSH levels and increasing lipid ROS levels [112]. To sum up, reducing the levels of GSH may be feasible to induce ferroptosis in tumor cells.
5.1.4. Ferroptosis Inducers Targeting GPX4
GPX4 converts L-OOH into non-toxic L-OH, preventing ferroptosis [32]. ML-162 induces ferroptosis in thyroid cancer cells by reducing the activity of GPX4 (Table 5 [113]. RSL3, a reagent containing electrophilic chloroacetamide, induces ferroptosis on DLBCL and renal cell carcinoma in vitro in a dose- and time-dependent manner through a decrease in the expression of GPX4 [29].
5.1.5. Ferroptosis Inducers Targeting DHODH
Dihydroorotate DHODH is reported to defend against ferroptosis, which operates dependent on mitochondrial GPX4 to inhibit ferroptosis in the IMM via reducing ubiquinone to ubiquinol, a radical-trapping antioxidant with anti-ferroptosis activity. In mitochondria, DHODH and GPX4 constitute two major ferroptosis defense systems by reducing the damage of LPO to mitochondria. Based on that, brequinar has been reported to damage the ferroptosis defense systems of low GPX4 tumors whose GPX4 defense system has low defense capability to inhibit their growth by inhibiting mitochondrial DHODH [96].
5.1.6. Multiple Targeted Ferroptosis Inducers
Some molecules induce ferroptosis through the joint action of the multiple targets above, including GPX4, SLC7A11, system Xc−, etc. Alloimperatorin induces breast cancer cell ferroptosis by reducing mRNA and protein expression levels of SLC7A11 and GPX4, which promotes the accumulation of Fe2+, ROS, and MDA, thereby inhibiting cell growth and invasion (Table 5) [114]. Bupivacaine, previously used as a local anesthetic, has recently been reported to modulate ferroptosis in bladder cancer by amplifying the level of Fe2+ and restraining the expression of system Xc− and GPX4 [115].
5.2. Ferroptosis-Based Combinational Cancer Therapy
Combining some of the above molecules will have a better therapeutic effect. Chemical hybridization of sulfasalazine and DHA promotes the death of brain tumor cells [116]. Combined treatment with brequinar and sulfasalazine can induce ferroptosis, suppressing GPX4′s high tumor growth [96].
5.3. Nanomedicines Treat Tumors via Inducing Ferroptosis
With better targetability in inducing ferroptosis by recombination with other substances, new nanoparticles have been developed to treat tumors. IpGdIO-Dox, a novel magnetic nanocatalyst set through iRGD-PEG-ss-PEG-modified gadolinium engineering magnetic iron oxide-loaded Dox, is reported for MRI-guided chemo- and ferroptosis synergistic cancer therapies. Specifically, ipGdIO-Dox induces cancer ferroptosis by releasing abundant Fe2+ ions and then catalyzing H2LPCA into highly toxic OH•, which damages mitochondria and cell membranes [118].
6. Conclusions
In conclusion, as a form of regulatory cell death, ferroptosis mainly depends on iron-mediated oxidative damage and subsequent cell membrane damage. The key to inducing ferroptosis is to increase iron accumulation, free radical production, fatty acid supply, and lipid peroxidation. In recent years, ferroptosis has shown exciting prospects in tumor treatment. A large number of known drugs and molecules have been proven to be able to treat ferroptosis. The treatment strategies of ferroptosis have also begun to diversify, which brings hope for tumor treatment.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241210037
This entry is offline, you can click here to edit this entry!