Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Diffuse large B-cell lymphoma (DLBCL) is aggressive, highly heterogeneous, and the most common form of B-cell lymphoma. Based on cell-of-origin algorithm category and oncogenic mechanisms, DLBCL is classified into three subsets: germinal center B cell-like, activated B cell-like, and unclassifiable subtype. Tumor cells have made some metabolic changes to protect themselves from malnutrition. These metabolic alterations affect the tumor microenvironment and macroenvironment.
- diffuse large B-cell lymphoma
- metabolic alteration
- metabolic regulation
- immune environment
1. Introduction
Cells take up various nutrients and undergo different metabolic processes to maintain their growth and differentiation. In normal and tumor cells, they utilize nutrients in different ways. For tumor cells, even when oxygen is present, glucose is still catalyzed by a series of enzymes to generate lactate, rather than being completely oxidized to carbon dioxide, which is called aerobic glycolysis (also known as the Warburg effect). Metabolisms of fatty acids and amino acids have also changed dramatically in tumor cells. These cell-intrinsic metabolic alterations together lead to a highly acidic, nutrient-poor and hypoxic tumor microenvironment (TME), which aggravates the processes of metabolic reprogramming in tumor cells and immune cells within the TME [1]. Ultimately, the metabolic reprogramming accelerates the formation of a tumor-promoting macroenvironment, which is considered a characteristic of cancer [2].
Diffuse large B-cell lymphoma (DLBCL) is aggressive, highly heterogeneous, and the most common form of B-cell lymphoma. Based on cell-of-origin algorithm category and oncogenic mechanisms, DLBCL is classified into three subsets: germinal center B cell-like, activated B cell-like, and unclassifiable subtype [3]. Based on consensus cluster classification with specific metabolic features, DLBCL is classified into three subsets: oxidative phosphorylation (OXPHOS), B-cell receptor/proliferation, and host response subtype [4]. This classification considers more heterogeneous features of DLBCL except for genomics and distinguishes different tumor types from a metabolic perspective [5].
Tumors, including B-cell malignancies, have developed a variety of metabolic strategies to evade antitumor immunity [2]. For instance, DLBCL cells reprogram glucose, amino acid, and fatty acid metabolisms to protect themselves from malnutrition, and subsequently immune cells force themselves to develop metabolic adaptations. However, these metabolic adaptations might reduce the antitumor effectiveness of immune cells [1][6][7][8][9][10]. Developing drugs that regulate cell metabolism could provide a good direction for cancer therapy [11].
2. Metabolic Alterations of DLBCL Macroenvironment
The concept of “tumor macroenvironment” was introduced by Al-Zoughbi W et al. in 2014 to indicate the pathological interaction among tumor cells, tumor microenvironment and other systems of the body [12]. It is reported that some soluble factors released by tumor cells affect the microenvironment and then reflect on the macroenvironment. Changes in the macroenvironment in turn promote tumor development. Some release soluble factors, such as chemokines, cytokines, or growth factors, which can not only recruit inflammatory cells (such as fibroblasts and myeloid cells) to the microenvironment but also remodel extracellular matrix, initiate and support the formation of new blood vessels, which support tumor growth and lead to more aggressive properties of tumor cells. In addition, new blood vessel networks, nearby tumor cells, and inflammatory cells are imperfect, which leads to accumulation of soluble factors in the tumor microenvironment and then cause pathological endocrine effects and the interaction of the tumor microenvironment with the patient’s organ system [13][14]. Increased levels of inflammatory mediators such as IL-6, TNF-α, IL-1, and IFN-γ are detected in the macroenvironment and are associated with systemic inflammatory responses. Some investigations have demonstrated that the multifactorial in situ network of inflammation controls the complex signaling processes that contribute to tumorigenesis and progression [15][16][17][18]. However, the mechanistic interaction between macroenvironmental factors and tumor development needs to be further explored.
Distinct metabolic features in peripheral plasma were investigated in DLBCL patients with different prognostic outcomes. Mi M et al. collected pretreatment serum samples from 80 DLBCL patients, including germinal center B cell-like (GCB) subtypes and non-GCB subtypes. Then, they tested the serum by the gas chromatography–mass spectrometry (GC-MS) technique, and identified valine, hexadecenoic acid, and pyroglutamic acid as the most important altered metabolites for the prognosis of DLBCL. Higher levels of pyroglutamic acid and hexadecenoic acid in serum are associated with better survival, while increased valine is associated with worse survival [19]. Another team analyzed plasma metabolomics from 22 healthy controls, 25 newly diagnosed DLBCL patients, and 18 complete-remission DLBCL patients. The results demonstrated that compared with complete-remission patients, newly diagnosed patients had decreased levels of glucose and aspartate in the plasma [20].
In addition, several studies have reported that abnormal immunologic markers in autoimmune diseases are significantly associated with the prognosis of DLBCL, which indicates these substances affect disease development through the macroenvironment. Chronic active BCR signaling interacts with several kinase pathways and contributes to DLBCL development, while lack of LYN kinase leads to an autoimmune disease [18][21]. Patients that have high levels of rheumatoid factors, such as anti-double-stranded DNA IgG and anti-nuclear antibody in serum, tend to experience a recurrence of DLBCL [22][23][24]. Abnormal serum immunoglobulins and complements are significantly associated with poor prognosis of patients. For example, there is increased serum IgE level or decreased serum complement C3 level in patients with high International Prognostic Index (IPI) score or high Eastern Cooperative Oncology Group (ECOG) score [25]. Although it remains unclear how tumor cells affect the tumor microenvironment and then change the tumor macroenvironment, this suggests that as tumors expand or disappear, the macroenvironment metabolism changes simultaneously.
3. Metabolic Alterations of Tumor Cells in DLBCL Microenvironment
3.1. Altered Glucose Metabolism
To meet energy demands, cells utilize glucose-derived carbon through glycolysis or OXPHOS. Although these two metabolic processes occur simultaneously, different types of cells have preferences to use certain metabolic axes. Low-proliferating or highly differentiated cells primarily depend on OXPHOS under aerobic conditions and turn on glycolysis under hypoxia. However, tumor cells, as highly proliferating cells, attain most adenosine triphosphate (ATP) through aerobic glycolysis, even when oxygen is present. This aerobic glycolysis can lead to increased glucose uptake and lactate generation [26]. 18F-FDG PET-CT, which is short for 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography–computed tomography, has been widely utilized in diagnostics and assessment of the therapeutic response in B-cell lymphoma [27]. Several studies have indicated that increased FDG uptake has relationships with expression levels of hexokinase 2 (HK2), glucose transporters (GLUTs) and monocarboxylate transporters (MCTs) in DLBCL tumor cells [28][29].
HK2 is a vital member of the HK family, as the first key rate-limiting enzyme to facilitate glucose to glucose-6-P (G6P) during glycolysis process. It is reported that overexpression of HK2 increases tumor cell glycolysis, and a high level of HK2 is relevant to poor prognosis in DLBCL patients [30]. GLUTs, glucose transporters, can carry and transport glucose across plasma membrane. Their expression and localization are regulated by some factors. For example, nuclear translocation of NF-κB drives transcription of a gene encoding a scaffold that allows AKT phosphorylation to increase GLUT1 localization to the plasma membrane, while tumor protein P53 (TP53), MYC and hypoxia-inducible factor-1α (HIF-1α) upregulate the expression levels of GLUTs and thus lead to greater glycolytic flux in DLBCL [31][32]. MTCs can transfer lactate between different cell types, and are essential for the tumor microenvironment due to high levels of lactate produced by anaerobic glycolysis. Patients with MCT1 upregulation in tumor cells and MCT4 upregulation in stromal cells tend to have poor prognosis. In MYC-amplified lymphomas, MCT1 expression was dramatically increased, which indicates MCT1 expression is regulated by MYC [33][34][35] (Figure 1).
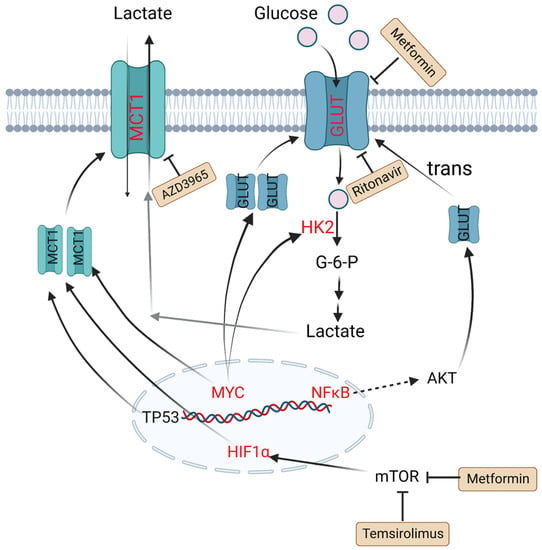
Figure 1. Altered glucose metabolism and potential drugs in DLBCL. Nuclear translocation of NF-κB drives transcription of a gene encoding a scaffold that allows AKT phosphorylation to promote GLUT1 localization to the plasma membrane. MYC promotes lactate production by upregulating HK2 and increases GLUT synthesis. TP53, HIF1α and MYC upregulate the expression level of MCT1. Metformin and temsirolimus are mTOR inhibitors and ritonavir blocks glucose uptake by binding to GLUT4. AZD3965 is an inhibitor of MCT1. Molecules in red represent upregulation in DLBCL. PI3K: phosphoinositide 3-kinase; GLUT: glucose transporter; NF-κB: nuclear factor kappa-B; TP53: tumor protein P53; HIF1α: hypoxia-inducible factor-1α; mTOR: mammalian target of rapamycin; HK2: hexokinase 2; MCT1: monocarboxylate transporter-1.
3.2. Altered Amino Acid Metabolism
In tumor cells, changes in amino acid metabolism can promote tumor development and progression. For amino acid utilization, tumor cells prefer glutamine as a useful energy source [36]. Glutamine is converted into glutamate by glutaminase (GLS) and finally generates ATP through the tricarboxylic acid (TCA) cycle. It is reported that in many tumor cell lines including B lymphoma, the metabolism of glutamine is modulated by MYC via increasing the expression of GLS. The glutamine transporter protein expression of solute carrier family 1 member 5 (glutamine transporter SLC1A5, also called ASCT2) and solute carrier family 7 member 5 (SLC7A5) can be upregulated by MYC to increase the uptake of glutamine [37]. NAD-dependent deacetylase sirtuin-3 (SIRT3) is a mitochondrial protein deacetylase that stimulates glutamine dissolution by activating GDH to increase α-ketoglutarate (α-KG) production. High expression of SIRT3 in DLBCL tumor cells can replenish the TCA cycle and generate ATP to create biomass, which supports tumor growth [38].
Except for glutamine metabolism, other amino acid metabolisms also play important roles in DLBCL. Indoleamine 2,3 dioxygenase (IDO) is a rate-limiting enzyme that catalyzes tryptophan to kynurenine (Kyn) and kynurenic acid [39]. These two products are considered immunosuppressive, since they are agonists of the aryl hydrogen receptor (AHR). AHR induces Treg generation and supports tumor growth by regulating oncogene expression [8]. It is reported that in DLBCL patients, a third of cases have positive IDO expression and worse prognosis compared with IDO-negative DLBCL patients. Sun C et al. found inhibition of IDO1 restrained DLBCL cell proliferation, leading to the upregulation of TP53 in RNA seq analysis [40]. Protein arginine methyltransferase-5 (PRMT5) is an essential enzyme to catalyze dimethylation of arginine on diverse cytoplasmic or nuclear substrates (such as histones) and enhances immunosuppressive function [41]. Erazo T et al. reported that MYC may act as a key target of PRMT5 in B-cell lymphomas [42]. The hexosamine biosynthesis pathway (HBP) requires glucose and glutamine for the O-linked N-acetylglucosamine (O-GlcNAc) cycle, which plays an important role in posttranslational protein modification that adds GlcNAc to nuclear and cytoplasmic proteins. A recent study indicated that the substrates of HBP and O-GlcNAc transferase (OGT) are upregulated in DLBCL tumor cells. Consumption of glucose and glutamine or treatment with an HBP inhibitor (azaserine) in DLBCL cells can reduce the O-GlcNAc substrates and inhibit the activation of NF-κB, further inducing G0/G1 cell arrest and apoptosis [43] (Figure 2).
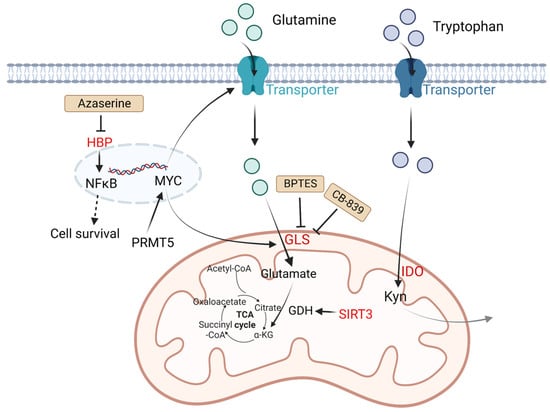
Figure 2. Altered amino acid metabolism and potential drugs in DLBCL. Glutamine is transported by glutamine transporters and then converted into glutamate by GLS. SIRT3 stimulates glutamine dissolution by activating GDH to increase α-KG production, which leads to enhancement of the TCA cycle and thereby generates more ATP to support tumor growth. IDO is a rate-limiting enzyme that catalyzes tryptophan to Kyn and kynurenic acid, which are considered immunosuppressive. PRMT5 promotes transcription of MYC. MYC activates GLS and glutamine transporters. HBP activates the NF-κB signaling pathway. The HBP inhibitor azaserine in DLBCL cells can inhibit the activation of NF-κB, and thereby induce cell arrest and apoptosis. BPTES is a selective allosteric regulator of GLS1 that attenuates the growth of lymphoma xenografts and delays MYC-driven tumor proliferation. Another GLS inhibitor, CB-839 inhibits the conversion of glutamine to glutamate. Kyn, catalyzed by IDO1 from tryptophan, is considered immunosuppressive. Molecules in red represent upregulation in DLBCL. GLS: glutaminase; SIRT3: NAD-dependent deacetylase sirtuin-3; PRMT5: protein arginine methyltransferase-5; HBP: the hexosamine biosynthesis pathway; ATP: adenosine triphosphate; TCA: tricarboxylic acid; GDH: glutamate dehydrogenase; α-KG: α-ketoglutarate; Kyn: kynurenine; NF-κB: nuclear factor kappa-B.
3.3. Altered Fatty Acid Metabolism
To meet increased energy needs, tumor cells reprogram fatty acid metabolism to achieve rapid transformation and progression. The basic source of carbons for fatty acid synthesis in tumor cells comes from glucose. In the mitochondria, glucose is converted into acetyl-CoA then citrate. The mitochondrial citrate transporter protein (CTP) transfers citrate out of the mitochondria. Subsequently, cytosolic citrate and coenzyme A (CoASH) are catalyzed by ATP citrate lyase (ACLY) to oxaloacetate and acetyl-CoA, and the latter is used to synthesize fatty acids [44].
Fatty acid synthase (FASN) is an important enzyme to synthesize 16-carbon fatty acid palmitate, which is highly expressed in more than half of DLBCL samples. Its overexpression is related to poor prognosis. Its inhibitor fasnall is a selective FASN inhibitor that targets multiple domains of FASN, which can restrain c-Met receptor kinase and promote apoptosis of DLBCL cells [45]. The transmembrane protein CD36 functions as the cell surface channel for exogenous fatty acid uptake, which is upregulated and strongly correlated with the proportion of M2 macrophages in CD5+ DLBCL. [46].
As a tumor suppressor protein, phosphatase and tensin homologue (PTEN) alterations are found in about 10% of DLBCL cell lines [47], which have a significant opposite relationship with FASN expression in prostate cancer [48]. PTEN dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3), while phosphoinositide 3-kinase (PI3K) phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate PIP3 [49]. Clinically, a majority of PIK3CA mutations are found in PTEN+ cases, which indicates a significant correlation between PTEN and the PI3K/AKT signaling pathway [50] (Figure 3).
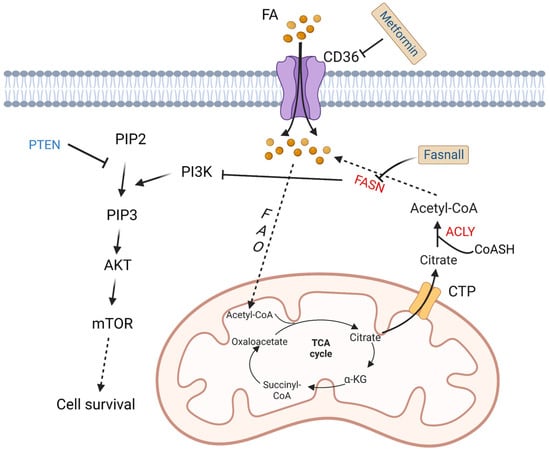
Figure 3. Altered fatty acid metabolism and potential drugs in DLBCL. Fatty acids are transported into DLBCL cells via CD36. Fatty acids are finally converted to acetyl-CoA by a series of oxidation reactions and enter the TCA cycle. A product in the TCA cycle, citrate is transported out of mitochondria by CTP. In cytoplasm, citrate and CoASH are converted to acetyl-CoA catalyzed by ACLY. FASN, the key enzyme in fatty acid synthesis, inhibits the activation of PI3K. PI3K phosphorylates PIP2 to PIP3, and then activates AKT/mTOR pathway to promote cell survival. On the contrary, PTEN dephosphorylates PIP3. Fasnall is a selective FASN inhibitor. Metformin is an inhibitor of CD36. TCA: tricarboxylic acid; FAO: fatty acid oxidation; FASN: fatty acid synthesis; α-KG: α-ketoglutarate; ACLY: ATP-citrate lyase; PYR: pyruvate; CTP: citrate transporter protein; PTEN: phosphatase and tensin homologue; mTOR: metabolic regulator mammalian target of rapamycin; CoASH: coenzyme A. PIP3: phosphatidylinositol 3,4,5-trisphosphate; PIP2: phosphatidylinositol 4,5-bisphosphate.
This entry is adapted from the peer-reviewed paper 10.3390/metabo13060734
References
- Lian, X.; Yang, K.; Li, R.; Li, M.; Zuo, J.; Zheng, B.; Wang, W.; Wang, P.; Zhou, S. Immunometabolic rewiring in tumorigenesis and anti-tumor immunotherapy. Mol. Cancer 2022, 21, 27.
- Ravi, S.; Alencar, A.M., Jr.; Arakelyan, J.; Xu, W.; Stauber, R.; Wang, C.I.; Papyan, R.; Ghazaryan, N.; Pereira, R.M. An Update to Hallmarks of Cancer. Cureus 2022, 14, e24803.
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858.
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861.
- Jalali, S.; Ansell, S.M. The potential role of glycogen metabolism in diffuse large B-cell lymphoma. Leuk. Lymphoma 2020, 61, 1028–1036.
- Pallasch, C.P.; Leskov, I.; Braun, C.J.; Vorholt, D.; Drake, A.; Soto-Feliciano, Y.M.; Bent, E.H.; Schwamb, J.; Iliopoulou, B.; Kutsch, N.; et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 2014, 156, 590–602.
- Hude, I.; Sasse, S.; Engert, A.; Bröckelmann, P.J. The emerging role of immune checkpoint inhibition in malignant lymphoma. Haematologica 2017, 102, 30–42.
- Beielstein, A.C.; Pallasch, C.P. Tumor Metabolism as a Regulator of Tumor-Host Interactions in the B-Cell Lymphoma Microenvironment-Fueling Progression and Novel Brakes for Therapy. Int. J. Mol. Sci. 2019, 20, 4158.
- Pi, M.; Kuang, H.; Yue, C.; Yang, Q.; Wu, A.; Li, Y.; Assaraf, Y.G.; Yang, D.-H.; Wu, S. Targeting metabolism to overcome cancer drug resistance: A promising therapeutic strategy for diffuse large B cell lymphoma. Drug. Resist. Updat. 2022, 61, 100822.
- Turturro, F. Constitutive NF-κB Activation Underlines Major Mechanism of Drug Resistance in Relapsed Refractory Diffuse Large B Cell Lymphoma. BioMed Res. Int. 2015, 2015, 484537.
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033.
- Al-Zoughbi, W.; Al-Zhoughbi, W.; Huang, J.; Paramasivan, G.S.; Till, H.; Pichler, M.; Guertl-Lackner, B.; Hoefler, G. Tumor macroenvironment and metabolism. Semin. Oncol. 2014, 41, 281–295.
- Dieterich, L.C.; Bikfalvi, A. The tumor organismal environment: Role in tumor development and cancer immunotherapy. Semin. Cancer Biol. 2019, 65, 197–206.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444.
- Argilés, J.M.; Busquets, S.; López-Soriano, F.J. Cytokines in the pathogenesis of cancer cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 401–406.
- Staal-van Den Brekel, A.J.; Dentener, M.A.; Schols, A.M.W.J.; Buurman, W.A.; Wouters, E.F.M. Increased resting energy expenditure and weight loss are related to a systemic inflammatory response in lung cancer patients. J. Clin. Oncol. 1995, 13, 2600–2605.
- Kayacan, O.; Karnak, D.; Beder, S.; Güllü, E.; Tutkak, H.; Senler, F.Ç.; Köksal, D. Impact of TNF-alpha and IL-6 levels on development of cachexia in newly diagnosed NSCLC patients. Am. J. Clin. Oncol. 2006, 29, 328–335.
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92.
- Mi, M.; Liu, Z.; Zheng, X.; Wen, Q.; Zhu, F.; Li, J.; Mungur, I.D.; Zhang, L. Serum metabolomic profiling based on GC/MS helped to discriminate Diffuse Large B-cell Lymphoma patients with different prognosis. Leuk. Res. 2021, 111, 106693.
- Fei, F.; Zheng, M.; Xu, Z.; Sun, R.; Chen, X.; Cao, B.; Li, J. Plasma Metabolites Forecast Occurrence and Prognosis for Patients With Diffuse Large B-Cell Lymphoma. Front. Oncol. 2022, 12, 894891.
- Chan, V.W.; Meng, F.; Soriano, P.; DeFranco, A.L.; Lowell, C.A. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 1997, 7, 69–81.
- Ekström Smedby, K.; Vajdic, C.M.; Falster, M.; Engels, E.A.; Martínez-Maza, O.; Turner, J.; Hjalgrim, H.; Vineis, P.; Seniori Costantini, A.; Bracci, P.M.; et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: A pooled analysis within the InterLymph Consortium. Blood 2008, 111, 4029–4038.
- Baecklund, E.; Iliadou, A.; Askling, J.; Ekbom, A.; Backlin, C.; Granath, F.; Catrina, A.I.; Rosenquist, R.; Feltelius, N.; Sundström, C.; et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 692–701.
- Bilici, A.; Yapici, H.S.; Ercan, S.; Seker, M.; Ustaalioglu, B.B.; Salman, T.; Orcun, A.; Gumus, M. The prevalence and significance of autoantibodies in patients with non-Hodgkin’s lymphoma: Are they correlated with clinicopathological features? J. BUON 2012, 17, 502–507.
- Sheng, L.; Fu, D.; Cao, Y.; Huo, Y.; Wang, S.; Shen, R.; Xu, P.; Cheng, S.; Wang, L.; Zhao, W. Integrated Genomic and Transcriptomic Analyses of Diffuse Large B-Cell Lymphoma With Multiple Abnormal Immunologic Markers. Front. Oncol. 2022, 12, 790720.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Kobe, C.; Kuhnert, G.; Kahraman, D.; Haverkamp, H.; Eich, H.T.; Franke, M.; Persigehl, T.; Klutmann, S.; Amthauer, H.; Bockisch, A.; et al. Assessment of tumor size reduction improves outcome prediction of positron emission tomography/computed tomography after chemotherapy in advanced-stage Hodgkin lymphoma. J. Clin. Oncol. 2014, 32, 1776–1781.
- Schöder, H.; Noy, A.; Gönen, M.; Weng, L.; Green, D.; Erdi, Y.E.; Larson, S.M.; Yeung, H.W.D. Intensity of 18fluorodeoxyglucose uptake in positron emission tomography distinguishes between indolent and aggressive non-Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 4643–4651.
- Elstrom, R.; Guan, L.; Baker, G.; Nakhoda, K.; Vergilio, J.-A.; Zhuang, H.; Pitsilos, S.; Bagg, A.; Downs, L.; Mehrotra, A.; et al. Utility of FDG-PET scanning in lymphoma by WHO classification. Blood 2003, 101, 3875–3876.
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Author Correction: Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci. Rep. 2018, 8, 7221.
- Sommermann, T.G.; O’Neill, K.; Plas, D.R.; Cahir-McFarland, E. IKKβ and NF-κB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res. 2011, 71, 7291–7300.
- Soleja, M.; Mims, M.; Rivero, G. Uncovering molecular abnormalities leading to the Warburg effect in primary refractory diffuse large B-cell lymphoma. Blood Cancer J. 2016, 6, e502.
- Afonso, J.; Pinto, T.; Simões-Sousa, S.; Schmitt, F.; Longatto-Filho, A.; Pinheiro, C.; Marques, H.; Baltazar, F. Clinical significance of metabolism-related biomarkers in non-Hodgkin lymphoma–MCT1 as potential target in diffuse large B cell lymphoma. Cell. Oncol. (Dordr.) 2019, 42, 303–318.
- Doherty, J.R.; Yang, C.; Scott, K.E.N.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920.
- Gan, L.; Xiu, R.; Ren, P.; Yue, M.; Su, H.; Guo, G.; Xiao, D.; Yu, J.; Jiang, H.; Liu, H.; et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene 2016, 35, 3037–3048.
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121.
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765.
- Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239.
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy–Challenges and Opportunities. Trends Pharm. Sci. 2018, 39, 307–325.
- Ninomiya, S.; Hara, T.; Tsurumi, H.; Hoshi, M.; Kanemura, N.; Goto, N.; Kasahara, S.; Shimizu, M.; Ito, H.; Saito, K.; et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann. Hematol. 2011, 90, 409–416.
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress. 2020, 4, 199–215.
- Erazo, T.; Evans, C.M.; Zakheim, D.; Chu, K.L.; Refermat, A.Y.; Asgari, Z.; Yang, X.; Da Silva Ferreira, M.; Mehta, S.; Russo, M.V.; et al. TP53 mutations and RNA-binding protein MUSASHI-2 drive resistance to PRMT5-targeted therapy in B-cell lymphoma. Nat. Commun. 2022, 13, 5676.
- Pham, L.V.; Bryant, J.L.; Mendez, R.; Chen, J.; Tamayo, A.T.; Xu-Monette, Z.Y.; Young, K.H.; Manyam, G.C.; Yang, D.; Medeiros, L.J.; et al. Targeting the hexosamine biosynthetic pathway and O-linked N-acetylglucosamine cycling for therapeutic and imaging capabilities in diffuse large B-cell lymphoma. Oncotarget 2016, 7, 80599–80611.
- Park, J.K.; Coffey, N.J.; Limoges, A.; Le, A. The Heterogeneity of Lipid Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 33–55.
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Ajarim, D.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol. Cancer Ther. 2010, 9, 1244–1255.
- Liu, M.K.; Cheng, L.L.; Yi, H.M.; He, Y.; Li, X.; Fu, D.; Dai, Y.T.; Fang, H.; Cheng, S.; Xu, P.P.; et al. Enhanced lipid metabolism confers the immunosuppressive tumor microenvironment in CD5-positive non-MYC/BCL2 double expressor lymphoma. Front. Oncol. 2022, 12, 885011.
- Sakai, A.; Thieblemont, C.; Wellmann, A.; Jaffe, E.S.; Raffeld, M. PTEN gene alterations in lymphoid neoplasms. Blood 1998, 92, 3410–3415.
- Bandyopadhyay, S.; Pai, S.K.; Watabe, M.; Gross, S.C.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Saito, K.; Markwell, S.J.; et al. FAS expression inversely correlates with PTEN level in prostate cancer and a PI 3-kinase inhibitor synergizes with FAS siRNA to induce apoptosis. Oncogene 2005, 24, 5389–5395.
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front. Oncol. 2014, 4, 252.
- Abubaker, J.; Bavi, P.P.; Al-Harbi, S.; Siraj, A.K.; Al-Dayel, F.; Uddin, S.; Al-Kuraya, K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia 2007, 21, 2368–2370.
This entry is offline, you can click here to edit this entry!