Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Fluorescent proteins (FPs) are optical probes that are used to track the functions of genetically encoded target molecules in molecular and cellular biology. FPs have intrinsic photophysical properties generated by the chromophore and its surrounding amino acid sequences. The intensity of the fluorescence emission of FPs can be changed using external factors such as pH or metal ions.
- fluorescent protein
- fluorescent quenching
- FP
1. Introduction
Metal ions are essential for all living organisms and play critical roles in fundamental biological processes such as osmotic regulation, catalysis, metabolism, biomineralization, and cell signaling [1]. Lack of essential metal ions in an organism can cause growth disorders, severe disruption in body functions, carcinogenesis, or death [2]. Moreover, a deficiency or excess of metal ions in living organisms can cause health problems, and their concentrations must be maintained within appropriate ranges for optimal cellular function [3,4,5,6]. Therefore, investigating the distribution and concentration fluctuations in metal ions within a cell can provide valuable insights for medical diagnoses related to cell signaling, metabolic engineering, or disease tracking [7,8].
Transition metal ions such as Cu, Ni, and Zn are involved in several physiological and pathophysiological pathways [9]. Several spectrophotometric and electroanalytical techniques, such as atomic absorption/emission spectroscopy [10,11], inductively coupled plasma mass spectroscopy [12,13,14], electrochemical assays [15,16], and colorimetric methods [17,18], have been widely used and have provided reliable results. However, these methods have several limitations in terms of expensive equipment, complex preprocessing, detection speed, and immediate detection in the field [7]. Therefore, the development of more accessible metal-detection probes is required. Among the various potential probe materials, fluorescent probes are attractive for metal detection because of their promising photophysical properties such as high sensitivity and selectivity, low detection limit, fast response, operational simplicity, real-time monitoring, and low cost [7,19].
In the field of biology, various biomolecules, such as peptides [20], enzymes [21], antibodies [22], nucleic acids [23], DNAzymes [24], and whole cells [25], are being developed as metal biosensors to measure metal ion levels. Fluorescent proteins (FPs) have photophysical properties because they emit fluorescence when the light of a specific wavelength is absorbed [26,27,28,29]. FPs are widely used as optical probes to track the location and function of genetically encoded target molecules in various molecular and cell biology techniques, such as Förster or fluorescence resonance energy transfer (FRET) [30,31,32], biosensors [33,34,35,36,37], optogenetics [38,39,40], chemogenetics [41,42], subcellular localization [43,44,45], in vivo imaging [46,47,48], or genome editing [49,50,51,52] (Table 1).
Table 1. Application of fluorescent proteins in biological research.
| Application | Biological Research | Reference |
|---|---|---|
| Förster or fluorescence resonance energy transfer (FRET) | Protein-protein interactions or conformational changes within proteins | [30,31,32] |
| Biosensors | Monitoring of small biomolecules or other physiological intracellular processes | [33,34,35,36,37] |
| Optogenetics | Measuring or controlling molecular signals, cells, or groups of cells | [38,39,40] |
| Chemogenetics | Monitoring cellular receptors that affect signal pathways within a cell | [41,42] |
| Subcellular localization | Monitoring the location of the target molecule in cells | [43,44,45] |
| In vivo imaging | Imaging plasmids or protein-protein interactions in organs | [46,47,48] |
| Genome editing | Monitoring genome editing | [49,50,51,52] |
Green fluorescent protein (GFP) was first discovered in jellyfish (Aequorea victoria) [53]. GFP-like proteins form a β-can fold comprising an 11-stranded β-sheet [54]. A peptide portion penetrates the β-barrel, and the center of the β-barrel contains a chromophore composed of a tripeptide (Figure 1).
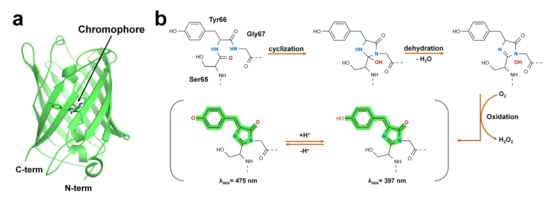
Figure 1. Structure of green fluorescent protein and chromophore. (a) Crystal structure of the green fluorescent protein (GFP) from jellyfish (PDB code: 1EMA). (b) GFP chromophore formation following a three-step autocatalytic process including cyclization, dehydration, and oxidation of three amino acids (Ser65, Tyr66, Gly67). Absorption properties of GFP are dependent on chromophore protonation states resulting in two absorption maxima in the excitation spectrum.
The FP chromophore is generated by post-translational modifications through processes such as folding, cyclization, dehydration, and oxidation [26] (Figure 1b). The chromophore of FP includes two or more ring structures, which are the most important sites for the optical properties of fluorescent proteins [55]. The quantum efficiency and absorption and emission wavelengths of FPs are affected by the constituting amino acids of the chromophore and the microenvironment surrounding the chromophore [26,55]. In addition, hydrogen bonds around the chromophore of FPs or bridges with water molecules are formed, and the amino acids surrounding the chromophore are involved in determining the unique optical properties of FPs [27,28,56]. According to their absorption and emission wavelengths, FPs can be classified into blue/UV, cyan-green, yellow, orange, red, and far-red colors (Table 2).
Table 2. Classification of fluorescent proteins based on the fluorescence emission wavelength.
| Color | Protein | λEx (nm) | λEm (nm) | QY | Oligomerization | Reference |
|---|---|---|---|---|---|---|
| Blue/UV | Sirius | 355 | 424 | 0.24 | Monomer | [57] |
| Azurite | 383 | 447 | 0.55 | Weak dimer | [58] | |
| moxBFP | 385 | 448 | 0.56 | Monomer | [59] | |
| Cyan | Aquamarine | 430 | 474 | 0.89 | Monomer | [60] |
| Cerulean | 433 | 475 | 0.62 | Weak dimer | [61] | |
| CyPet | 435 | 477 | 0.51 | Weak dimer | [62] | |
| mTurquoise2 | 434 | 474 | 0.93 | Monomer | [63] | |
| Green | mEGFP | 488 | 507 | 0.60 | Monomer | [64] |
| mClover3 | 506 | 518 | 0.78 | Monomer | [65] | |
| mNeonGreen | 506 | 517 | 0.80 | Monomer | [66] | |
| Yellow | mGold | 515 | 530 | 0.64 | Monomer | [67] |
| mCitrine | 516 | 529 | 0.74 | Monomer | [64] | |
| mVenus | 515 | 527 | 0.64 | Monomer | [68] | |
| Orange | mOrange | 548 | 562 | 0.69 | Monomer | [69] |
| mKO2 | 551 | 565 | 0.62 | Monomer | [70] | |
| TurboRFP | 553 | 574 | 0.67 | Dimer | [71] | |
| tdTomato | 554 | 581 | 0.69 | Tandem dimer | [69] | |
| Red | mApple | 568 | 592 | 0.49 | Monomer | [72] |
| mScarlet | 569 | 594 | 0.70 | Monomer | [73] | |
| mCherry | 587 | 610 | 0.22 | Monomer | [69] | |
| DsRed2 | 561 | 587 | 0.55 | Tetramer | [74] | |
| Far-Red | mPlum | 590 | 649 | 0.10 | Monomer | [75] |
| mRaspberry | 598 | 625 | 0.15 | Monomer | [75] | |
| mNeptune | 600 | 650 | 0.20 | Monomer | [76] | |
| TagRFP657 | 611 | 657 | 0.10 | Monomer | [77] |
Moreover, based on their unique optical properties and applications, FPs can be classified into large Stokes shifts, photoactivatable, photoswitchable, and photoconvertible FPs (Table 3).
Table 3. Classification of fluorescent proteins based on optical properties.
| Property | Protein | λEx (nm) | λEm (nm) | QY | Brightness | Oligomerization | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Large Stokes Shift | tKeima | 440 | 616 | 0.22 | 3.19 | Tetramer | [79] | |
| LSSmKate2 | 460 | 605 | 0.17 | 4.42 | Monomer | [80] | ||
| T-sapphire | 399 | 511 | 0.60 | 26.4 | Weak dimer | [81] | ||
| Photoactivatable | PAmKate | 586 | 628 | 0.18 | 4.5 | Monomer | [82] | |
| PAmCherry1 | 564 | 595 | 0.46 | 8.28 | Monomer | [83] | ||
| PATagRFP | 562 | 595 | 0.38 | 25.08 | Monomer | [84] | ||
| Photoswitchable | Dronpa | 503 | 518 | 0.85 | 80.75 | Monomer | [85] | |
| mGeos-M | 503 | 514 | 0.85 | 43.86 | Monomer | [86] | ||
| rsTagRFP | 567 | 585 | 0.11 | 4.05 | Monomer | [87] | ||
| Photoconvertible | Kaede | Green | 508 | 518 | 0.88 | 86.94 | Tetramer | [88] |
| Red | 572 | 580 | 0.33 | 19.93 | ||||
| mEos3.2 | Green | 507 | 516 | 0.84 | 53.26 | Monomer | [89] | |
| Red | 572 | 580 | 0.55 | 17.71 | ||||
| Dendra | Green | 492 | 508 | 0.65 | 58.5 | Tetramer | [90] | |
| Red | 557 | 575 | 0.68 | 23.8 | ||||
All FP exhibit unique and intrinsic fluorescence properties. However, these properties are altered by various external environments. Accordingly, FPs have been suggested as biosensors for intracellular calcium indicators [91], chloride indicators [34], pH indicators [36,56], and ligand monitoring of receptors [33]. In particular, FPs are characterized by fluorescence quenching when exposed to a specific metal ion (Figure 2) and can potentially be used as an attractive probe for metal biosensors.
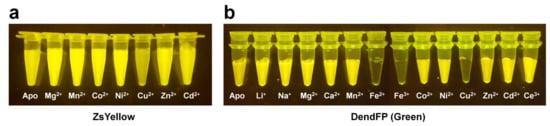
Figure 2. Visualization of quenchable metal ion screening for fluorescent proteins. Fluoresce quenching of (a) ZsYellow and (b) DendFP by metal ions.
The fluorescence quenching of FPs mediated by metal ions can be caused by static quenching [94], energy transfer between chromophores and quenchable metal ions [95], or perturbations in the FP structure [96]. To develop FP-based metal biosensors, it is important to understand the previous spectroscopic results for the fluorescence quenching of FP mediated by metal ions and the metal binding mechanism on FP.
2. Spectroscopic Properties of Metal-Induced FP
2.1. DsRed
DsRed (DrFP583) is a red fluorescent protein cloned from reef corals [101]. Eli and Chakrabartty investigated the fluorescent emission changes in response to metal ions in DsRed and its mutant proteins (gRF, Rmu74, Rmu80, Rmu162, and Rmu13) [97]. DsRed, Rmu13, and gRF showed a high percentage of quenching of Cu2+, whereas the mutants Rmu74, Rmu80, and Rmu162 showed modest quenching of Cu2+, indicating that the quenching effect of the metal can differ among the mutants despite using red fluorescence of the same origin. Rmu13 was characterized by dual-fluorescence emission peaks at 500 and 580 nm. The titration of DsRed (blue- and red-shifted mutants) and Rmu13 with Cu2+ yielded binding constants of 15 and 11 mM, respectively. Moreover, Sumner et al. reported that copper in the binding constant of DsRed is 0.5 mM [102], which differs from that reported by Eli and Chakrabartty. This apparent discrepancy is due to differences in the formulas for calculating the binding constants, which indicates the need for formalization of the binding constant calculation method applied when developing FP-based metal biosensors (see Section 3 Discussion). The dissociation constant (Kd) of the copper-binding site of DsRed and Rmu13 was approximately 14.80 and 10.90 μM, respectively.
Although structural evidence for quenchable metal binding to DsRed has not been reported yet, biochemical and spectral analyses clearly show a Cu2+-binding site on DsRed. Eli and Chakrabartty investigated copper binding after chemical modification using iodoacetamide and diethyl pyrocarbonate (DEPC) of cysteine and histidine residues, respectively, to trace the Cu2+-binding site of DsRed [97]. As a result, Kd increased by >100 μM when treated with iodoacetamide, whereas no remarkable difference was observed upon treatment with DEPC [97]. This result indicates that the cysteine residue of DsRed is involved in Cu2+-binding [97]. Rahimi et al. investigated the mechanism of DsRed fluorescence quenching: far-UV CD spectral analysis showed no structural changes in the entire protein, even when DsRed was bound to a metal ion [103]. In addition, the UV-visible spectra and Stern–Volmer constant showed a static quenching mechanism during the interaction between DsRed and Cu2+ [103]. The crystal structure of DsRed showed a tetramer with 222 non-crystallographic symmetry [104] (Figure 3a).
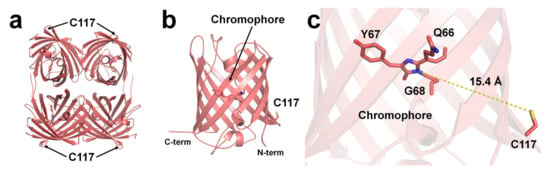
Figure 3. The Cu2+-binding site of DsRed (PDB code 1GGX). The position of Cu2+-binding Cys117 residue on the surface of β-can of (a) tetrameric and (b) monomeric DsRed. The Cu2+-binding site is identified based on chemical modification and spectral analysis. (c) The average distance between the imidazoline ring of the chromophore and Cys117 in DsRed was 15.4 Å.
To understand the Cu2+-binding site of DsRed, the deposited crystal structure of DsRed (PDB code 1GGX) was analyzed. The Cys117 residue, which can bind Cu2+ of DsRed, was located on the β7-strand of the β-can surface (Figure 3a,b). The closest distance between the sulfhydryl of Cys117 and the imidazolinone ring of the chromophore was approximately 15.4 Å (Figure 3c). Because the DsRed monomer contains only one cysteine residue, the tetrameric DsRed can bind four Cu2+ ions (Figure 3a). Moreover, as Cys117 of DsRed was located on the surface of β-can, the chelator can easily remove the Cu2+ bound to Cys117, which is consistent with the previous spectral results [97].
2.2. BFPms1
BFPms1 is an engineered fluorescent protein that uses a specific metal to bind GFP and alter its fluorescence signal [35]. To create the chromophore of BFPms1 as a metal ligand, such as porphyrin, Tyr66 (corresponding to the chromophore) was substituted with a histidine residue. In particular, Barondeau et al. have developed an H148G mutant to generate holes between β-strands for metal access (Figure 4a). Moreover, additional amino acid substitutions were performed to increase quantum yield (Y145F), improve solubility (F64L, F99S, M153T, and V163A), and promote rapid chromophore formation (S65T). The metal ion screening results showed that Cu2+ quenched the fluorescence emission whereas Zn2+ enhanced it. BPFms1 has a Kd of 24 μM and 50 μM for Cu2+ and Zn2+, respectively, whereas KD is greater than 2 mM for other transition metal ions [35]. The crystal structures of BFPms1 complexed with Zn2+ and Cu2+ were determined at a resolution of 1.44 and 1.50 Å, respectively. In Zn-bound BFPms1 (PDB code 1KYS), Zn2+ exhibited a trigonal bipyramidal geometry distorted by the chromophore Glu222 and water molecules (Figure 3b). In Cu-bound BFPms1 (1KYR), a square-planar geometry was exhibited by the amino acid Glu222 and the porphyrin of the chromophore (Figure 3c). The histidine ring of BFPms1 exhibited a metal bond shift and rearrangement of Glu222. Structural changes around the chromophore have been shown to occur depending on the type of metal ion bonded to the chromophore. However, it has not been clarified whether the fluorescence emission is changed by the conformational change of the chromophore or whether a metal is bound to the chromophore and has an effect. In this review, additional metal ions binding to the surface of β-can from coordinates deposited in PDB (PDB code 1KYR and 1KYS) were observed. In the BFPms1-Zn2+ structure, three additional Zn2+ ions interacted with the His25, Glu142, and Glu172 residues, whereas in BFPms1-Cu2+, Mg2+ ions interacted with Glu142 in the final model structure. In a structural study of BFPms1, only the metal around the chromophore was described [35], and further studies are required to determine whether the metal bound to the surface of BFPms1 affects fluorescence emission.
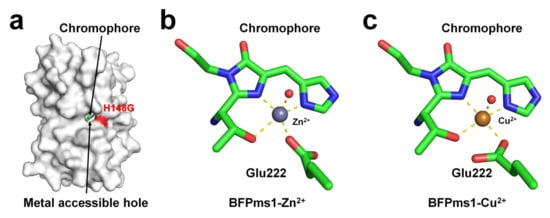
Figure 4. Crystal structure of native, Zn-bound (PDB code 1KYS), and Cu-bound (PDB code 1KYR) state of BFPms1. (a) Surface structure of BFPms1 showed an engineered hole, where the metal ion is accessible. (b) Zn2+ and (c) Cu2+ binding to the chromophore of BFPms1.
2.3. iq-mEmerald
Two surface-exposed histidine residues separated by one residue of the β-sheet (i and i+2) create robust transition metal ion binding sites in proteins [95,105]. Using this protein engineering concept, Yu et al. have generated mutant constructs of mEmerald-1H (His147), mEmerald-2H (His202 and His204), and mEmerald-3H (His147, His202, and His204). In mEmerald-2H, two histidine residues are located on the same β10-strand [9]. In Emerald-3H, the His147 residue is located at the β7-strand, which is the closest neighbor position of the two histidine residues of mEmerald-2H. mEmearld-2H and mEmerald-3H showed strong fluorescent quenching for Cu2+ with a Kd of 0.3 and 0.2 μM, respectively, whereas wild-type mEmerald and mEemrald-1H showed weak (at high concentration) or low affinity (1–10 μM), respectively. Accordingly, Yu et al. have generated the most robust and sensitive mEmerald-3H mutant (iq-mEmerald) for Cu2+-induced fluorescence quenching. Physiological concentrations of Ca2+ (1 mM) and Mg2+ (10 mM) did not affect the fluorescence quenching behavior of iq-mEmerald. The engineered histidine motif of iq-mEmerald can bind specifically to other transition metal ions, such as Co2+, Ni2+, and Zn2+. The crystal structure of iq-mEmerald complexed with Ni2+ (PDB code 4KW8) and Zn2+ (4KW9) ions were determined at a resolution of 2.45 Å, and 1.80 Å, respectively (Figure 5a). Both Ni2+ and Zn2+ interacted with His202 and His204 of iq-mEmerald; however, the conformations of the side chains of the metal-interacting histidine residues differed (Figure 5b). In native iq-mEmerald, the side chains of His202 and His204 were in the direction opposite to the metal-binding site. In iq-mEmerald-Ni2+, the side chains of His202 and His204 were rotated toward the metal-binding site, and Ni2+ was coordinated by His202 and His204 at a distance of 2.08 and 2.04 Å, respectively. In contrast, in iq-mEmerald-Zn2+, the conformation of His202 was similar to the conformation of His202 in metal-free iq-mEmerald, whereas the side chain of His204 was rotated to Zn2+ ion. Zn2+ was coordinated by His202 and His204 at a distance of 2.01 and 2.00 Å, respectively. The closest distance between the imidazoline ring and metal-binding site for Ni2+ and Zn2+ was approximately 15.7 and 13.4 Å, respectively (Figure 5c). This result indicates that the metal-binding configuration and the distance between the metal ion and chromophore differ depending on the type of metal ion, although the metal-binding sites are the same.
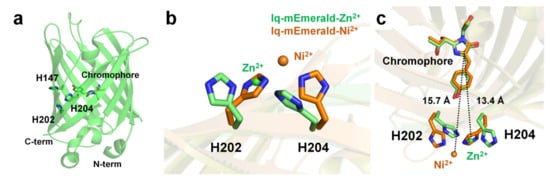
Figure 5. Crystal structure of the native and metal-bound states of iq-mEmerald. (a) Engineered sites (H147, H202 and H204) of iq-mEmerald for metal binding. (b,c) Comparison of the metal-binding site between apo (green) and nickel-bound (orange) structures.
In contrast, in iq-mEmerald-Zn2+ (PDB code 4KW8) deposited in PDB, one Cl− was modeled at a position 2.24 Å away from Zn2+ interacting with His202 and H204. Moreover, in the final model structure of iq-mEmerald-Zn2+, four Zn2+ were observed, which interacted with Glu124, His139, His147, and His169. However, the effect of the addition of metal ions has not yet been described in related structural studies [9]. Accordingly, it is necessary to investigate the metal-binding sites observed at nonengineered locations and further study their effects on fluorescence emission. This structural information is useful for understanding the metal-binding moiety for further engineering of FP-based metal biosensors. In addition, because the crystal structure of iq-mEmerald complexed with the most effective quencher Cu2+ has not yet been determined, further crystallographic studies of Cu2+-bound iq-mEmerald are required to better understand the molecular mechanism of Cu2+ binding to iq-mEmerald.
2.4. Dronpa
Dronpa is a photoswitchable green fluorescent protein isolated from the Echinophyllia sp. SC22 [85]. The Dronpa chromophore consists of a CYG tripeptide with maximum excitation and emission wavelengths of 503 and 518 nm, respectively [85]. The cysteine residue of the Dronpa chromophore is critical for its photoswitchable function [106]. The optical properties of metal-induced fluorescence quenching of Dronpa were screened using divalent ions such as Mg2+, Ca2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, or Co2+ [98]. Cu2+ clearly exerted the strongest quenching effect on the fluorescence emission of Dronpa, and Co2+ reduced the fluorescence intensity, whereas other metal ions did not significantly affect the fluorescence intensity of Dronpa. The fluorescence intensity of Dronpa quenched by Cu2+ and Co2+ was reversible and recovered by 97% and 95%, respectively, upon the addition of an EDTA chelator. Dronpa naturally has three histidine residues (His194, His210, and His212) located on the surface of the β-barrel. The crystal structures of Dronpa complexed with Co2+, Ni2+, and Cu2+ had a resolution of 2.20, 1.90, and 2.85 Å, respectively. In the Dronpa-Co2+, the Co2+ interacted with the His194 and His212 residues at a distance of 2.21 and 2.26 Å, respectively, and showed distorted octahedral coordination along with four water molecules. In Dronpa-Ni2+, the Ni2+ interacted with the His194 and His212 residues at 2.12 and 2.19 Å, respectively, and showed distorted octahedral coordination along with four water molecules. Accordingly, the metal coordination features of Dronpa-Co2+ and Dronpa-Ni2+ were similar but distinct from those of the surrounding water molecules. The distance between Co2+/Ni2+ and the imidazoline ring of the chromophore was approximately 14.4 Å. In Dronpa-Cu2+, Cu2+ metal interacts with His210 and His212 at distances of 2.34 and 2.54 Å, respectively. However, the Cu2+-binding coordination was not clear because of the poor electron density map. The side chains of His210 and His212 were rotated to the Cu2+-binding site by 90° and 115°, respectively, compared with the native Dronpa structure. Accordingly, when Dronpa interacts with a metal, the histidine residue at the binding site undergoes a conformational rearrangement, indicating that the metal has a geometrically stable configuration. The distance between Cu2+ and the imidazoline ring of the chromophore is approximately 14.9 Å. In this study, the metals around the chromophore were described [98]; however, additional metal ions were present at the bottom of the β-barrel in the deposited crystal structure. In Dronpa-Co2+, -Cu2+, and -Zn2+, additional metal ions were bound to His200, and the side chain was exposed to the solvent (Figure 6a). Moreover, the distance between the imidazoline ring of the chromophore and His200 was approximately 19 Å. Accordingly, further experiments are required to determine whether the metal bound to His200 affects the fluorescence-quenching ability of Dronpa.
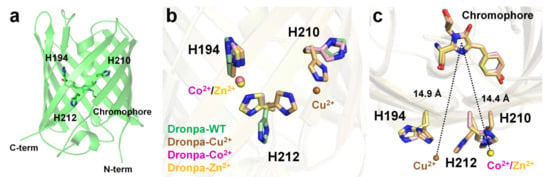
Figure 6. Crystal structure of native (PDB code 2GX2) and the Cu2+- (5HZT), Co2+-(5HZS), and Zn2+ (5HZU)-bound-state of Dronpa. (a) Dronpa contains three native histidine residues (H194, H210, and H212) on the β-can as the metal-binding site. (b) Comparison of the metal-binding site and the conformational change of histidine residues in Dronpa. (c) Measurement of distance between the metal ion and the chromophore of Dronpa.
This entry is adapted from the peer-reviewed paper 10.3390/chemosensors11040216
This entry is offline, you can click here to edit this entry!