1. Autofagia e cellule staminali AML
Durante lo sviluppo leucemico, le LSC possono adattare il loro metabolismo e i meccanismi autofagici per fornire l'elevata energia e i nutrienti necessari per la proliferazione e la sopravvivenza delle LSC in condizioni di carenza di nutrienti, fame, ipossia o durante i trattamenti chemioterapici [7
, 91
, 92
, 93
, 94
] .
La mitofagia, la funzione mitocondriale e l'integrità possono influenzare la vitalità, la proliferazione e il potenziale di differenziazione e la longevità delle normali cellule staminali ematopoietiche (HSC) [95]
e sono importanti nella strategia di sopravvivenza delle cellule staminali AML (LSC).
In particolare, la proteina chinasi (AMPK) attivata dall'adenosina5′-monofosfato (AMP), un complesso proteico fondamentale nel metabolismo mitocondriale e nella mitofagia, è costitutivamente attivata nelle LSC, aumentando la clearance mitocondriale per supportare la crescita e la sopravvivenza delle LSC attraverso il suo obiettivo a valle FIS1, la fissione mitocondriale 1 componente proteico di un complesso mitocondriale che promuove la fissione mitocondriale [
96 ]. La mitofagia mediata da AMPK/FIS1 è necessaria per l'auto-rinnovamento e la sopravvivenza delle LSC [
96]. Una sovraespressione di FIS1 è stata trovata anche nelle cellule AML mentre la deplezione di FIS1 compromette la mitofagia, indebolendo la capacità di auto-rinnovamento delle LSC e determinando l'induzione della differenziazione mieloide mediante l'inattivazione di GSK3 (glicogeno sintasi chinasi 3), indicando così la mitofagia come meccanismo di regolazione per la progressione della LMA [
96 ]. Più recentemente, la perdita del sequestosoma 1 (
SQSTM1 ), noto anche come p62, un recettore autofagico selettivo cruciale per lo sviluppo e la progressione dell'AML in vivo, induce l'accumulo di mitocondri danneggiati e superossido mitocondriale, compromettendo così la sopravvivenza delle cellule leucemiche. Quindi, la perdita di
SQSTM1 compromette la progressione della leucemia nei modelli murini di LMA, sottolineando il ruolo della mitofagia nella sopravvivenza delle LSC.
97 ]. Complessivamente, questi studi dimostrano che è necessaria una maggiore attività autofagica delle LSC per la progressione maligna in AML.
Tuttavia, contrariamente all'attivazione dell'autofagia osservata nell'AML, una perdita di autofagia nelle HSC sane innesca l'espansione di una popolazione di cellule progenitrici nel midollo osseo, dando origine a una mieloproliferazione grave e invasiva, come nell'AML umana [53
] . Questo apparente paradosso può essere spiegato dai ruoli distinti che l'autofagia può svolgere durante la progressione dell'AML, che può essere diversa nelle varie fasi della leucemogenesi [
98 ,
99]. L'autofagia nelle HSC normali può prevenire l'insorgenza del cancro, come meccanismo soppressivo del tumore. Infatti, l'autofagia rimuove gli organelli danneggiati, come i mitocondri, e protegge le cellule ematopoietiche dall'instabilità genomica e dall'infiammazione, prevenendo così l'insorgenza della leucemia. In particolare, l'aumento del danno al DNA, gli alti livelli di ROS, l'aneuploidia e un accumulo aberrante di p62/SQSTM1 sono stati correlati con un processo autofagico alterato, indicando un ruolo chiave dell'autofagia nella prevenzione dell'inizio del tumore [100
] . Al contrario, nel cancro accertato, l'autofagia può funzionare come un percorso favorevole che promuove la sopravvivenza e la crescita del tumore, aiutando le cellule tumorali a sfuggire allo stress metabolico e agli stimoli di morte.
Alcuni studi hanno anche dimostrato che l'attivazione del flusso autofagico gioca un ruolo solo nell'inizio dell'AML, con una trasformazione da HSC a LSC, e, quindi, dopo questa fase, l'autofagia non è necessaria per il mantenimento della malattia [101
] . Gli studi sull'AML MLL-AF9, l'alterazione più comune nell'AML infantile, indicano ATG5 o ATG7 come necessari per l'insorgenza dell'AML, ma una volta stabilita la condizione leucemica, l'autofagia non è richiesta per la funzione LSC in vivo [101
, 102
] . Tuttavia, in un diverso modello di topo AML MLL-ENL, il knockout Atg5 o Atg7 ha ridotto il numero di LSC funzionali, aumentato l'attivazione mitocondriale e i livelli di ROS in queste cellule e prolungato la sopravvivenza dei topi leucemici [103]
.]. In questo contesto, durante il processo di leucemogenesi, la metilazione dell'istone può regolare gli effettori autofagici centrali e i regolatori autofagici a monte come ATG 5 e ATG7 per influenzare indirettamente il livello di autofagia [
104 ]. Insieme, questi studi suggeriscono un ruolo altamente complesso e dipendente dal contesto per l'autofagia nella trasformazione leucemica rispetto alle proprietà di mantenimento delle LSC nell'AML.
Il duplice ruolo dell'autofagia nell'AML, come promotore o soppressore del cancro nell'AML, è ancora oggetto di dibattito. Gli studi hanno dimostrato che l'autofagia può agire come meccanismo pro o anti-proliferativo a seconda del lignaggio e del contesto genotipico molecolare della malattia, riflettendo il grado di eterogeneità dell'AML [105
] .
2. Regolazione dei geni dell'autofagia nelle cellule AML
Numerosi studi hanno dimostrato che l'aumento dell'autofagia nelle cellule AML conferisce protezione dal trattamento chemioterapico e promuove la sopravvivenza delle cellule AML.
L'aumentata autofagia mediata da ATG7 è stata associata a scarsi risultati clinici e a una breve durata della remissione nei pazienti con LMA [
106 ]. Più recentemente, alcune proteine coinvolte nella sopravvivenza delle cellule leucemiche e sovraespresse nell'AML sono state correlate alla sovraespressione di ATG, alla base dell'interazione tra autofagia e sovraespressione proteica che promuove la sopravvivenza delle cellule leucemiche [8]
.]. Hu et al. hanno dimostrato che un'elevata espressione di SIRT1 (Sirtuin 1), un attore chiave nella biogenesi mitocondriale e nella proteina correlata all'autofagia, è associata a un'elevata espressione di CXCR4, un marker prognostico negativo nell'AML e ad altre proteine correlate all'autofagia come ATG5 e LC3 in campioni umani primari di AML, indicando un ruolo potenziale della via di segnalazione SDF-1α-CXCR4 nell'induzione dell'autofagia nelle cellule AML, che promuove ulteriormente la loro sopravvivenza sotto stress [107
] .
Il canale ionico del recettore transitorio della melastatina 2 (TRPM2), coinvolto nel mantenimento della sopravvivenza cellulare dopo il danno ossidativo, è altamente espresso nell'AML [
108 ]. Eseguendo l'esaurimento
di TRPM2 , Chen SJ et al. [
108 ] hanno dimostrato che i livelli di proteine ULK1, Atg7 e Atg5 sono diminuiti nelle cellule impoverite
di TRPM2 , portando all'inibizione dell'autofagia. È importante sottolineare che l'esaurimento di
TRPM2 nell'AML inibisce la proliferazione della leucemia e aumenta la sensibilità alla doxorubicina delle cellule AML [
108 ].
Studi funzionali su normali cellule CD34 + CB hanno indicato che l'inibizione dell'espressione di VMP1 ha ridotto il flusso autofagico, con diminuzione dell'espansione delle cellule staminali e progenitrici ematopoietiche (HSPC), differenziazione ritardata, aumento dell'apoptosi e compromissione della funzione cellulare e dell'attecchimento in vivo. Risultati simili sono stati osservati nelle linee cellulari leucemiche e nelle cellule AML CD34+ primarie. Inoltre, l'analisi ultrastrutturale ha indicato che le cellule leucemiche che sovraesprimono VMP1 hanno un numero ridotto di strutture mitocondriali e il numero di strutture di degradazione lisosomiale è aumentato. La sovraespressione di VMP1 (vacuole membrane protein-1) ha aumentato il flusso autofagico e migliorato la qualità mitocondriale, che ha coinciso con un aumento della soglia per la perdita indotta da venetoclax della permeabilizzazione della membrana esterna mitocondriale (MOMP) e dell'apoptosi nelle cellule leucemiche.
109 ].
Delezioni eterozigoti, mutazioni missenso o cambiamenti nel numero di copie dei geni chiave dell'autofagia sono stati trovati con un'alta frequenza nei pazienti con LMA, in particolare nei pazienti con LMA con cariotipi complessi [5
, 103
] . In particolare, una perdita cromosomica eterozigote di 5q, 16q o 17p è correlata alle regioni che codificano i geni autofagici
ATG10 e
ATG12 ,
GABARAPL2 e
MAP1LC3B o
GABARAP , rispettivamente [
103 ], e molti altri geni autofagici hanno un basso livello di espressione nell'AML umano blasti, un flusso autofagico ridotto e alti livelli di ROS [
103 ]. Inoltre, uno studio ha suggerito che i geni chiave dell'autofagia come
ULK1 ,
ATG3 ,
ATG4D e
ATG5 erano significativamente sottoregolati nelle cellule AML primarie rispetto ai normali granulociti [
110 ].
3. Biomarcatori autofagici
Recentemente sono stati compiuti progressi significativi per identificare specifici geni correlati all'autofagia per la previsione degli esiti clinici nell'AML. Insieme ai geni
ATG precedentemente descritti, diversi microRNA implicati nella leucemogenesi e nella chemioresistenza sono stati coinvolti anche nell'attivazione dell'autofagia e possono essere utilizzati come biomarcatori [
111 ]. In particolare, la sovraespressione di miR-17-5p nella leucemia promuove la proliferazione di AML inibendo l'autofagia attraverso il targeting BECN1 [
112 ,
113 ,
114 ]. Gansan et al. dimostrato che le cellule stromali sottoregolano i livelli di miR-23a-5p nelle cellule leucemiche, portando alla sovraregolazione dell'autofagia protettiva in queste cellule, aumentando così la loro resistenza alla tossicità della chemioterapia.
115 ]. È stato dimostrato che la sovraespressione di MiR-143 migliora la sensibilità delle cellule AML alla citotossicità del trattamento con citarabina (Ara-C) inibendo l'autofagia attraverso il targeting ATG7 e ATG2B [
116 ]. Una sovraespressione di miR-15a-5p è coinvolta nella chemioresistenza dei pazienti con LMA, attraverso i geni correlati all'autofagia
ATG9A ,
ATG14 ,
GABARAPL1 e
SMPD1 che prendono di mira le cellule AML [
117 ].
I recenti progressi nella bioinformatica hanno prodotto una firma correlata all'autofagia che può aiutare a prevedere la sopravvivenza globale (OS) e/o gli esiti clinici dei pazienti con LMA. Diversi studi hanno dimostrato che la progressione dell'AML dipende dalla firma genica associata all'autofagia [
118 ]. Un recente studio di bioinformatica ha costruito un modello contenente 10 geni correlati all'autofagia per prevedere la sopravvivenza dei pazienti affetti da AML, dimostrando che i gruppi ad alto rischio di AML hanno una maggiore espressione di geni del checkpoint immunitario e una percentuale più alta di cellule CD4 T e NK [119]
.]. Inoltre, questo studio è stato in grado di predire l'OS nella LMA attraverso la firma di 10 geni, indicando questo modello come un efficace predittore prognostico per i pazienti con LMA, utile per guidare la stratificazione dei pazienti per immunoterapie e farmaci [119
] . Lo studio di bioinformatica regressione LASSO Cox che ha identificato una firma di rischio critico per AML, costituito dai geni autofagici
BAG3 ,
CALCOCO2 ,
CAMKK2 ,
CANX ,
DAPK1 ,
P4HB ,
TSC2 e
ULK1 , aveva un eccellente potere predittivo per la prognosi AML [
120]. In particolare, le citochine immunosoppressive sono risultate significativamente aumentate nel microambiente tumorale di pazienti con un alto rischio di AML, previsto sulla base della firma correlata all'autofagia di questi pazienti [120
] . Tuttavia, il valore prognostico della firma ATG in ambito clinico è ancora dibattuto. Pertanto, i ruoli della firma ATG e dell'autofagia nella patogenesi dell'AML dovrebbero essere ulteriormente studiati.
Inoltre, uno studio interessante ha indicato che una firma di lncRNA correlata all'autofagia contenente sei lncRNA (
HYMAI ,
MIR155HG ,
MGC12916 ,
DIRC3 ,
C1orf220 e
HCP5 ) può avere un importante valore prognostico [
121 ]. Uno studio recente ha indicato quattro lncRNA associati all'autofagia (
MIR133A1HG ,
AL359715.1 ,
MIRLET7BHG e
AL356752.1 ) come firma da utilizzare potenzialmente come biomarcatore per predire la sopravvivenza dei pazienti con LMA [
122 ].
Complessivamente, questi dati indicano che il ruolo dell'autofagia nello sviluppo del tumore dipende chiaramente dal tipo di LMA e dallo stadio di sviluppo del tumore. Inoltre, l'autofagia può fornire alle cellule tumorali una strategia di sopravvivenza, suggerendo un uso terapeutico per l'inibizione dell'autofagia. D'altra parte, l'autofagia può indurre la morte cellulare, indicando l'attivazione dell'autofagia come una nuova strategia nella terapia del cancro. Pertanto, è necessario determinare il ruolo svolto dall'autofagia nei sottotipi molecolari di AML, o il grado di sviluppo del tumore, per verificare se la sua modulazione potrebbe portare a benefici per il paziente trattato.
4. Autofagia e alterazioni genetiche nell'AML
Il fenotipo AML deriva da molteplici alterazioni molecolari, genetiche ed epigenetiche che influenzano la differenziazione, la proliferazione e l'apoptosi dei progenitori mieloidi. L'Organizzazione Mondiale della Sanità ha classificato le AML in base alla presenza di particolari alterazioni genetiche, spesso originate da traslocazioni cromosomiche o altri riarrangiamenti del genoma come t(8;21), t(15;17), inv(16), inv(3) , t(6;9), t(9;11) o t(11;19), o mutazioni nelle chinasi del recettore, in mediatori di segnalazione chiave, proto-oncogeni o enzimi epigenetici, ad esempio, mutazioni in FLT3 (FMS
- like tirosina chinasi 3),
TP53 ,
c-KIT o
IDH1/2 ,
NPM1 (nucleofosmina 1) e proteina legante potenziatore CCAAT (
CEBPA ) [
1 ,
2 ,
123 ]. Queste mutazioni nelle AML hanno un impatto sulla scelta della terapia più adatta.
L'associazione tra autofagia e alterazioni genetiche ricorrenti è stata descritta in diversi studi sull'AML, ma necessita di ulteriori indagini [
124 ,
125 ]. Qui, riassumiamo e aggiorniamo i recenti progressi che hanno evidenziato il legame tra autofagia e geni di fusione e mutazioni oncogeniche ricorrenti nell'AML e il coinvolgimento dell'autofagia nel trattamento chemioterapico (
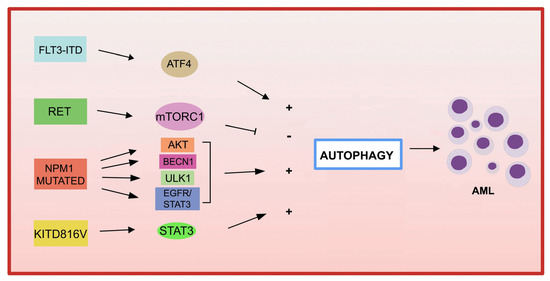
Figura 1 ).
Figura 1. Mutazioni genetiche nell'AML e nell'autofagia. Diverse anomalie genetiche ricorrenti nell'AML sono coinvolte nella deregolazione dell'autofagia, portando alla progressione della leucemia.
4.1. Geni di fusione in AML e autofagia
La maggior parte dei casi di APL sono causati da una traslocazione de novo t (15;17) (q22; q21), che risulta nella fusione del gene
RARA con il gene
PML [
126 ,
127 ]. Le cellule APL che hanno un'espressione inferiore di geni correlati all'autofagia rispetto alle cellule normali hanno un'attività autofagica ridotta. Utilizzando agenti differenzianti, come l'acido all
trans retinoide (ATRA) e il triossido di arsenico (ATO) attualmente utilizzati in ambito clinico, il livello di espressione dei geni correlati all'autofagia aumenta, ripristinando così l'autofagia nelle cellule APL [
128]. Entrambi gli agenti possono attivare l'ETosis, un tipo di morte cellulare mediata dal rilascio di trappole extracellulari di neutrofili (ET). Inoltre, l'azione autofagica dipendente da mTOR è necessaria per la NETosi indotta da ATO nelle cellule APL [
129 ]. Da notare che la rapamicina, l'inibitore della via mTOR, si sinergizza con l'ATO nell'eradicazione delle cellule che iniziano la leucemia (LIC) attraverso l'attivazione della NETosis sia nelle cellule APL che in un modello APL in vivo [129
] .
Le traslocazioni del gene della leucemia di linea mista (
MLL ) 11q23 sono state osservate in circa l'80% delle AML pediatriche. In questi, il gene
MLL può, mediante traslocazione genomica, essere fuso con >60 diversi partner di fusione [
130 ]. Il trattamento con l'inibitore dell'oncogene RAS, tipifamib, porta all'inibizione dell'AML con la traslocazione t(6;11) inducendo sia l'apoptosi che l'autofagia [
131 ]. Un altro studio ha dimostrato che l'ATG5 partecipa allo sviluppo della leucemia guidata da
MLL-AF9 , ma non nei topi chemioterapici sensibili all'AML che esprimono MLL-AF9 [
132 ].
La leucemia mieloide acuta con core binding factor (
CBF-AML ) è caratterizzata dalla presenza di t(8;21) (q22; q22), o inv(16) (p13q22)/t(16;16), che porta alla formazione di
RUNX1/RUNX1T1 (AML1/ETO) e
CBFbeta-MYH11 , rispettivamente [
133 ]. L'attivazione dell'autofagia mediata da ULK1 può controllare e ritardare la leucemogenesi guidata da
AML1-ETO9a in un modello murino knockout AML
CASPASE-3 [ 134 ], suggerendo che CASPASE-3 è un importante regolatore dell'autofagia nell'AML. I risultati di questi studi evidenziano i diversi ruoli dell'autofagia nell'inizio, nella progressione e nelle risposte chemioterapiche nelle cellule AML, a seconda del diverso tipo di oncoproteina aberrante.
4.2. Mutazioni genetiche in AML e autofagia
Tra le alterazioni genetiche più comuni nell'AML, la mutazione del gene della tirosina chinasi 3 ( FLT3 ) si verifica in circa il 30% dei casi di AML.
Le aberrazioni più frequenti che interessano il gene
FLT3 , associate a una prognosi sfavorevole nella LMA, sono la duplicazione tandem interna (
FLT3 -ITD) nel dominio juxtamembrana e le mutazioni puntiformi, che coinvolgono il dominio tirosin-chinasico di
FLT3 (
FLT3 -TKD) [
135 ] .
L'espressione di FLT3 -ITD aumenta l'autofagia basale nelle cellule AML attraverso un meccanismo che coinvolge il fattore di trascrizione ATF4 (attivazione del fattore di trascrizione 4) [
136 ]. Inoltre, l'inibizione dell'autofagia nelle cellule
FLT3 -TKD, che sono resistenti all'inibitore FLT3 quizartinib (AC220), inibisce anche la proliferazione sia in vitro che in vivo [
136]. Più recentemente, la mutazione D835Y acquisita ha indotto resistenza all'inibitore FLT3 sorafenib e ha attivato l'autofagia nelle linee cellulari
FLT3 -ITD-positive. Inibendo l'autofagia, gli autori sono stati in grado di superare la resistenza al sorafenib nella LMA
FLT3 -ITD-positiva, migliorandone l'efficacia [
137 ]. Recentemente, uno studio ha dimostrato che l'inibizione dell'autofagia riduce il potenziale di ripopolamento delle LSC
FLT3 -ITD AML associate all'accumulo mitocondriale [
138 ]. Inoltre, gli autori hanno dimostrato che l'inibizione dell'autofagia migliora l'attività di p53 e aumenta l'inibizione mediata da TKI dei progenitori dell'AML [
138 ].
L'autofagia non solo contribuisce alla proliferazione a valle del recettore FLT3-ITD, ma può anche essere coinvolta nella degradazione del recettore mutato. Infatti, in uno studio, è stata osservata la frequente attivazione del recettore tirosina chinasi RET in diversi sottotipi di LMA [
139 ]. RET media la soppressione dell'autofagia in modo dipendente da mTORC1, portando alla stabilizzazione del recettore FLT3 mutante. L'inibizione genetica o farmacologica di RET ha ridotto la crescita delle cellule AML dipendenti da FLT3, con la sovraregolazione dell'autofagia e la deplezione
di FLT3 [
139]. Questi risultati suggeriscono che il ripristino dell'autofagia nell'AML dipendente da FLT3 può comportare la degradazione dell'FLT3 mutato e quindi può rappresentare un interessante approccio terapeutico. È stato anche dimostrato che l'inibizione della proteina FLT3-ITD porta ad un aumento della sintesi di ceramide e media la mitofagia ceramide-dipendente, portando alla morte delle cellule AML [140
, 141
] .
Le mutazioni di KIT sono associate ad un'aumentata proliferazione di cellule leucemiche e ad un aumentato rischio di recidiva di AML [
142 ,
143 ]. Uno studio recente ha riportato che la mutazione
KIT D816V nelle cellule AML aumenta l'autofagia basale, stimolando la proliferazione e la sopravvivenza delle cellule AML tramite la segnalazione STAT3 [
144 ]. Una diversa mutazione puntiforme in c-
KIT (N822K T > A) attiva costitutivamente questo recettore, rendendo le cellule AML altamente sensibili al sunitinib (un inibitore della tirosina chinasi), con conseguente morte cellulare AML attraverso l'attivazione sia dell'apoptosi che dei processi autofagici [
145 ] .
Le mutazioni in
NPM1 (nucleophosmin 1) sono le alterazioni genetiche più frequenti nella LMA adulta, responsabili della localizzazione aberrante della proteina NPM1 nel citoplasma [
146 ]. L'aumentata attività autofagica riscontrata nelle cellule AML mutate con
NPM1 è coinvolta nella sopravvivenza delle cellule leucemiche [
147 ]. Il mutante
NPM1 può anche interagire con la proteina soppressore tumorale PML (proteina pro-mielocitica della leucemia), portando alla delocalizzazione e alla stabilizzazione della PML che, a sua volta, può attivare l'autofagia tramite la segnalazione AKT [147
] . In un altro studio, è stato dimostrato che nei pazienti affetti da LMA portatori di mutante
NPM1, l'enzima glicolitico PKM2 (piruvato chinasi M2) ha indotto l'autofagia attraverso la fosforilazione della proteina autofagica Beclin 1, contribuendo alla sopravvivenza cellulare [
148 ]. Infine, la proteina mutante NPM1 può anche interagire con la proteina autofagica ULK1, stimolando l'ubiquitinazione dipendente da TRAF6 di ULK1 tramite miR-146, mantenendo così la stabilità e la funzionalità di ULK1 e promuovendo la sopravvivenza delle cellule autofagiche [149
] . Inoltre, è stato osservato che l'espressione di RASGRP3, una proteina associata alla progressione del tumore, è sovraregolata nei pazienti con LMA con mutazione
NPM1 rispetto ai pazienti con LMA senza
NPM1 mutante . Gli autori hanno dimostrato che
NPM1-mut blocca la degradazione della proteina RASGRP3 attraverso il legame con la proteina MID1 dell'ubiquitina ligasi E3, portando alla sovraespressione di RASGRP3, oltre a promuovere l'attivazione a valle di EGFR-STAT3, che a sua volta promuove la proliferazione e l'autofagia nelle cellule AML [150
] .
Alterazioni del gene oncosoppressore
TP53 si riscontrano in circa il 5-15% dei casi di AML e, frequentemente, nei pazienti più anziani [
151 ,
152 ]. È stato proposto che il ruolo dell'autofagia nello sviluppo dell'AML possa essere determinato dallo stato di
TP53 . Per l'AML
TP53 wild-type , i ricercatori hanno dimostrato che il blocco farmacologico dell'autofagia ottiene benefici terapeutici, mentre le AML che ospitano mutazioni
TP53 non rispondono all'inibizione dell'autofagia da parte dell'idrossiclorochina (HCQ) [
153 ,
154 ]. L'uso di inibitori autofagici può essere una potenziale strategia terapeutica da utilizzare, in particolare per il trattamento di
TP53AML di tipo selvaggio. Per l'AML con mutazioni
TP53 , le vie autofagiche possono essere un'opzione terapeutica da utilizzare per l'eliminazione del mutante T
TP53 .
Un altro studio ha dimostrato che la stimolazione della macroautofagia da parte di 17-AAG, un inibitore HSP90, provoca la degradazione di TP53 R248Q nelle cellule AML e migliora anche la trascrizione dei geni associati all'autofagia [155
] . Inoltre, le prove accumulate indicano che TP53 attivato da una varietà di stress cellulari può innescare l'autofagia attraverso la transattivazione di geni pro-autofagici, tra cui
DRAM1 (modulatore autofagico regolato dal danno al DNA 1),
SESN1 (sestrin 1) e (sestrin
SESN2 ) [
155 ,
156 ,
157 ,
158 ].
Uno studio recente ha evidenziato il ruolo dell'autofagia nelle cellule AML, nel contesto dell'apoptosi mediata da p53, che è associata ad una maggiore citotossicità al trattamento con inibitori MDM2 e Ara-C quando il miR-10a è inibito [159
] . La strategia antileucemica basata sull'uso di inibitori MDM2/X nei tumori p53 wild-type per ripristinare la conformazione normale e attiva di p53, MDM2 e MDMX non è stata ampiamente testata [160
] . Pertanto, l'uso di una combinazione di trattamenti, inclusi gli inibitori MDM2 con modulatori autofagici, potrebbe essere una nuova strategia per migliorare il trattamento dell'AML
p53 wild-type .
I trattamenti farmacologici che modulano l'autofagia nei pazienti con LMA portatori di mutazioni
p53 partecipano alla degradazione delle proteine p53 aberranti. La mutazione puntiforme di
TP53 nel residuo aminoacidico R428 (R248Q), con attività di guadagno di funzione, dà origine ad attività maligna nelle cellule tumorali del polmone [
161 ] e una perdita della funzione di soppressione tumorale nell'AML [
162 ].
È interessante notare che il trattamento con l'inibitore Hsp90 (17-AAG) determina l'attivazione dell'autofagia mediata da chaperone, che induce la degradazione della proteina aberrante p53R248Q nelle cellule AML. In particolare, in condizioni di stress metabolico, 17-AAG induce l'interazione tra p53R248Q e la proteina chaperone Hsc70, innescando l'autofagia mediata da chaperone per degradare p53R248Q [
155 ]. Questi dati aprono nuove opportunità per studi futuri che potrebbero chiarire il coinvolgimento funzionale di diversi tipi di autofagia e la loro connessione con i meccanismi molecolari per migliorare le terapie antitumorali contro l'AML che ospita le diverse varianti
TP53 .I recenti progressi nella bioinformatica hanno consentito l'identificazione di diverse mutazioni epigenetiche che interessano l'AML, tra cui
IDH1/2 , Tet metilcitosina diossigenasi 2 (
TET2 ), DNA metiltransferasi 3A (
DNMT3A ) e
ASXL1 , tutte associate alla patogenesi dell'AML [
163 ,
164 ,
165]. Le proteine IDH sono isocitrato deidrogenasi, implicate in vari processi biologici, come il metabolismo energetico, la demetilazione dell'istone, la modificazione del DNA e l'adattamento all'ipossia. Sono necessari ulteriori studi per studiare terapie innovative basate sull'autofagia mirata in combinazione con l'ipometilazione del DNA per trattare le AML che ospitano determinati tipi di alterazioni epigenetiche.
Mutazioni nel gene
DNMT3A , un enzima coinvolto nella metilazione dei dinucleotidi CpG, sono presenti nel 20-23% dei pazienti adulti con AML de novo [
165 ]. Diversi studi hanno dimostrato che il trattamento di pazienti affetti da LMA con agenti che inibiscono la DNA metiltransferasi, come l'azacitidina (5-aza-2′-deossicitidina), induce l'attività autofagica nelle cellule di leucemia AML [166
] . Uno studio condotto su un modello murino knock-in condizionale
DNMT3A R878H, utilizzato per prevedere gli specifici RNA lunghi non codificanti (lncRNA) regolati dalle mutazioni
DNMT3A nell'AML, ha prima identificato 23
lncRNA espressi in modo differenziato , quindi i geni bersaglio a valle regolati da questi lncRNA , incluso
ATP6V1A, un gene critico correlato all'autofagia, la cui sovraespressione è associata a prognosi infausta nell'AML [
167 ]. Tuttavia, ci sono ancora poche prove di un coinvolgimento diretto delle mutazioni del gene
DNMT3A con attività autofagica nell'AML.
Sono necessari ulteriori studi per comprendere il significato funzionale dell'autofagia associata a diverse mutazioni genetiche nelle cellule AML.
This entry is adapted from the peer-reviewed paper 10.3390/cells12111553