Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Infertility is a global problem on the rise. The WHO defines it as a condition of the reproductive system that can be diagnosed when there is a “failure to achieve a clinical pregnancy after 12 months or more of regular unprotected sexual intercourse”. It occurs due to four broad causes: lifestyle choices, inheritable factors, health conditions, and aging, with a degree of overlap between each of these factors. There are various mechanisms and factors that contribute to infertility, most of which have some type of connection with oxidative damage.
- longevity
- aging
- oocytes
- reproduction
- fertility
- DNA
1. Metabolic and Molecular Mechanisms of Ovarian Aging
The number of oocytes present in females varies throughout life. During the first 5 months before birth, the numbers increase steadily to about 7 million. By the time of birth, the oocytes decrease to around 1 million through atresia [1][2]. Atresia is the natural process through which ovarian function is regulated. For a long time, follicular atresia has been hypothesised to be the primary cause of menopause and, thus, infertility [3]. Atresia works by activating the apoptotic pathways [4], leading to only around 400 oocytes maturing to the degree necessary for ovulation and fertilisation [4][5]. Working counter to atresia is the process of folliculogenesis; the mechanism that develops, matures and selectively releases oocytes to the uterus [6]. The balance of atresia to folliculogenesis is a complex and tightly controlled system. Atretogenic factors include tumour necrosis factor-α (TNF-α), Interleukin-6 and androgens [5]. Androgens, however, are also important for the normal functioning of oocytes in natural physiological concentrations. The PI3K pathway has two ovarian functions. First, when upregulated, it triggers the maturation of primordial follicles into primary follicles. Second, in low doses, it prevents atresia from occurring in the primordial follicle pool in its dormant state. Testosterone promotes the PI3K pathway in follicles, thus ensuring both the above crucial physiological functions can be maintained [7]. Androgens play an important role in stimulating atresia in further follicular stages to ensure mono-follicular activation in humans [8]. Finally, their production in theca cells also leads to the production of oestradiol, the primary female sex hormone, in granulosa cells [9]. There have been some attempts to use androgens for fertility treatment, which researchers explore but any attempts to disrupt the delicate balance that androgens have in the female reproductive system should be met with caution.
Folliculogenic factors include gonadotropins and downward intermediates such as Interleukin-1β, insulin-like growth factor-1 and oestrogens [5]. As age increases, besides the quantitative loss of oocytes, the risk of aneuploidy increases. Aneuploidy in oocytes is the unequal separation of chromosomes during meiosis I or II, resulting in gametes with less/more than a single set of 23 chromosomes [10].
2. General Factors Involved in Ovarian Aging
Ovarian aging mechanisms are still unclear, as is their effect on oocyte quality/quantity. It would, however, be beneficial to examine the established and novel mechanisms so that researchers may then theorize on how they may be targeted.
The rate at which atresia occurs is largely accelerated as a woman approaches menopause when compared to the more linear decline seen before the mid to late thirties [3][5]. For a long time, the modelling for ovarian follicular loss was thought to best fit a biphasic decrease [5] (Figure 1). This is because the rate of atresia seems to increase twofold in the late 30s (37–38) when a threshold of around 25,000 follicles is exceeded. If this modelling was correct, without the twofold increase, follicular depletion would not occur until around 70 years of age [5]. More recent evidence, however, suggests that the follicular decay more closely fits a power model of decay [11], suggesting a more gradual exponential decrease that may not occur at exactly 37 years old and without there being a particular follicle threshold that triggers accelerated recruitment.
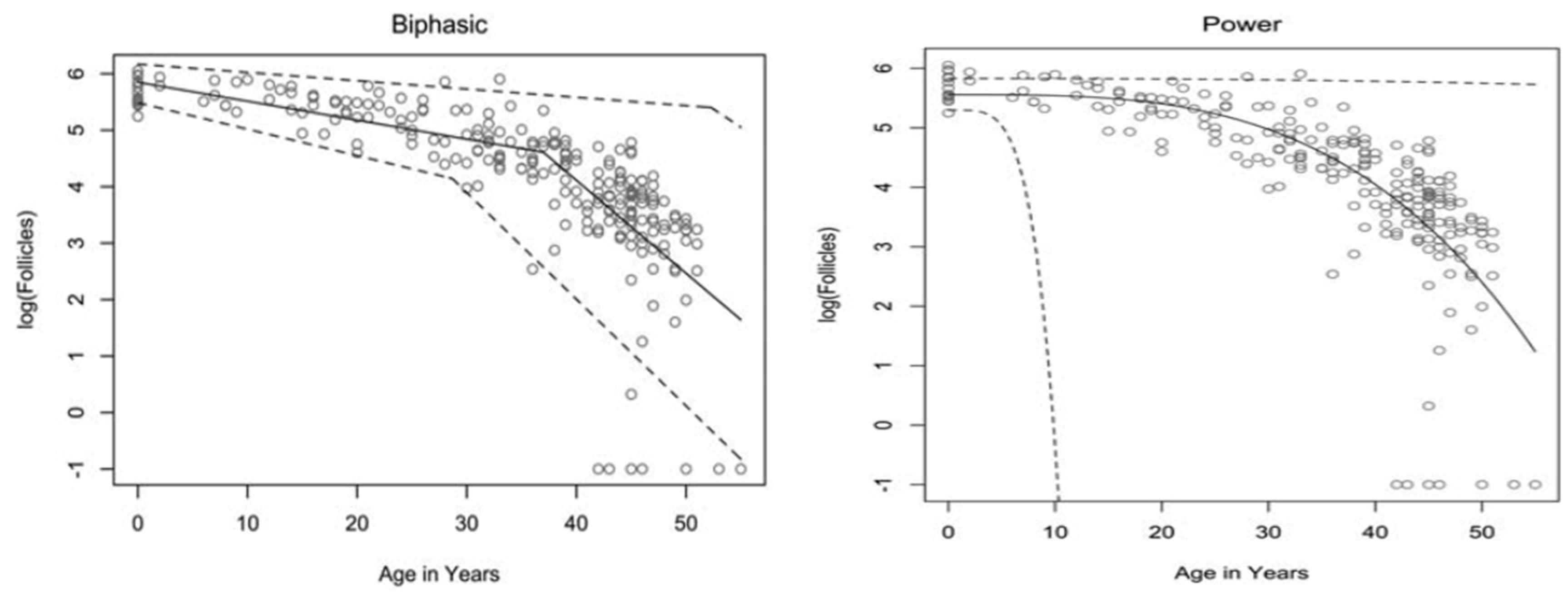
Figure 1. Models of human follicular loss over years, as seen in Coxworth et al. [11], with permission of Oxford University Press. Model represented by solid line, confidence intervals by dashed line.
2.1. Genetic Factors Involved in Ovarian Aging
The rate of atresia may be directly linked to the quality of the oocytes instead of the number of follicles. In mice that lack CHTF18, a protein that ensures chromatid cohesion and stable DNA replication, the rate of follicular atresia is notably higher than in CHTF18+ mice [12]. The increase in atresia also comes with an increase in aneuploidy and DNA double-strand breaks (DSB). Along the same lines, women with BRCA1 mutations experience earlier menopause along with higher aneuploidy [13][14]. This pattern is also seen in BRCA2 mutations [14], with primordial follicular density demonstrating a significant decrease compared to controls, especially after 30 years of age. The age of menopause is also linked to other genes related to homologous recombination such as BRE, MCM8 and INO80 [15].
It has been hypothesised that oocytes collect double-strand breaks in their DNA during their lifetime [16]. Titus et al. [16] showed that several genes involved in ATM-mediated DNA DSB repair pathways (BRCA1, ATM, MRE11, etc.) showed a decrease in expression after 37 years of age. However, the data were explained using a biexponential model of follicular decay [5] and, as discussed above, the decrease could be more gradual than the authors believe. The results could still fit into the more gradual power model (Figure 1). Researchers have seen above [13][14] that genetic BRCA1/2 mutations do lead to higher follicular atresia and a younger menopausal age. Taken together, the natural rate of atresia, which leads to menopause at around 45 years of age, may have the potential to be targeted for preventative treatment. If such treatment were successful, it would prolong menopause beyond this age limit and, thus, extend the female fertility window.
2.2. DNA Damage
DNA damage in the oocytes is accumulated over the reproductive lifespan. It may be caused by intrinsic factors, such as accrued replicated stress, ROS and hydrolysis, or by extrinsic factors, such as genotoxic substances, mutagens and radiation [17]. All these factors provide various mechanisms for damage incidents to occur on the oocyte DNA, whether structural or intrinsic. These can be thought of as “hits”, and when a certain number of hits is reached, the oocyte will be unsuitable for development into a foetus. It will then be either eliminated through apoptotic atresia or cause aneuploidy. The “hits” hypothesis [18] suggests that even if one mechanism for DNA damage is stopped or prevented, others will accumulate enough to still cause infertility. As they accumulate, they may have a domino effect, leading to exponential oocyte loss; the malfunction of one repair mechanism may have a detrimental effect on others, causing a cascade that tips over the delicate atresia/folliculogenesis equilibrium.
2.3. Cohesins
Cohesins are another factor leading to aging when they fail. They are the proteins that hold the sister chromatids together and are not renewed once formed; the same cohesins from foetal development must provide structural DNA support for up to 4 decades later [19][20]. After all, cohesins in oocytes sustain the sister chromatids in prophase I until the oocyte is selected for ovulation [20]. The fact that there is no cohesin turnover, combined with inexistent repair mechanisms, gives rise to the theory that cohesins play a vital role in causing aneuploidy [20], as sister chromatids separate prematurely. The “cohesin deterioration” hypothesis [21][22][23] suggests cohesion deterioration is the leading cause of aneuploidy, though the specific mechanism remains unclear [22]. It may be due to cohesin deterioration, primarily around the centromere [23]. Although cohesins also attach to specific points on the chromosomal arms [24], centromere cohesion appears to critically deteriorate first, with the mechanism for this deterioration remaining unclear [23].
2.4. Telomere Length
Telomere length may also influence oocyte quality. In a study with 120 POI patients, Xu et al. showed shorter telomere length in the granulosa cells compared to control [25]. Miranda-Furtando et al. [26], on a similar note, showed similar results in a study with 127 POI patients. Similarly, a significant difference in telomere length compared to control has been observed in patients with occult POI [27]. From these findings, it may be suggested that a short telomere length (and, therefore, reduced telomerase activity) may trigger atresia once a critical length has been reached. Additionally, though the cause of some forms of POI is still unknown, telomere length should be capable of acting as a predictive tool for the age of menopause and may furthermore be targeted as a mechanism to extend ovarian health for a longer time.
2.5. Oxidative Damage
A final factor is oxidative damage through deactivated antioxidative pathways. Ovarian aging has been shown to be linked to a downregulation of the pathways that prevent ROS from causing oxidative damage in both mitochondrial and nuclear DNA. On a non-human primate study, Wang et al. [28] demonstrated that a downregulation of transcription for key antioxidant mechanisms occurs with aging. This was shown by the statistically significant increase in DNA oxidation biomarker 8-OHdG and DNA damage biomarker γH2AX, among others. Such a study is important given that direct human ovarian samples are difficult to obtain, and it can provide a close model for ovarian aging. Oxidative damage may play a wider role and may be the predominant factor affecting all the mechanisms discussed above; it has been demonstrated to be linked to telomere shortening, genetic instability and subsequent apoptosis [29] through ovarian dysfunction (Figure 2). Furthermore, oxidative damage is shown to affect both mtDNA and DNA repair mechanisms [30]. In conclusion, the prevention of oxidative damage may be the key factor in preventing ovarian aging through the previously discussed pathways.
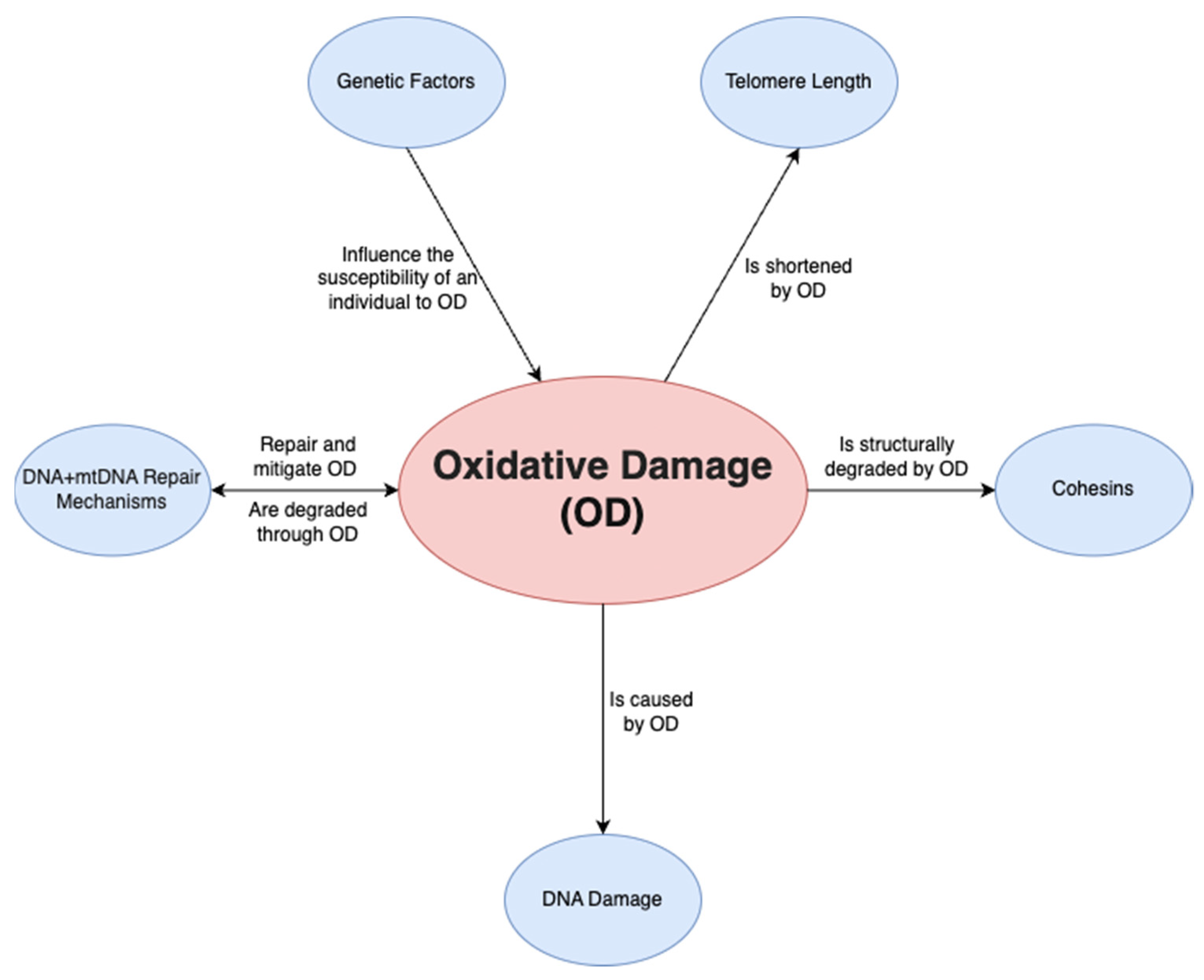
Figure 2. Relationship of oxidative damage to other mechanisms.
mtDNA is especially prone to DNA damage as it relies on the cell’s endogenous antioxidant mechanism rather than a mtDNA-specific mechanism for damage repair [31]. Mitochondria are in greater demand in oocytes compared to somatic cells, given the considerable ATP required for fertilisation and blastocyst formation. As such, mature (metaphase II) oocytes have up to 100,000 mitochondria [31], compared to the few thousand that may be found in somatic cells [32].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24129828
References
- Salha, O.; Abusheika, N.; Sharma, V. Dynamics of human follicular growth and in-vitro oocyte maturation. Hum. Reprod. Updat. 1998, 4, 816–832.
- Baker, T.G. A quantitative and cytological study of germ cells in human ovaries. Proc. R. Soc. London Ser. B Boil. Sci. 1963, 158, 417–433.
- Richardson, S.J.; Senikas, V.; Nelson, J.F. Follicular depletion during the menopausal transition: Evidence for accelerated loss and ultimate exhaustion. J. Clin. Endocrinol. Metab. 1987, 65, 1231–1237.
- Hansen, K.R.; Knowlton, N.S.; Thyer, A.C.; Charleston, J.S.; Soules, M.R.; Klein, N.A. A new model of reproductive aging: The decline in ovarian non-growing follicle number from birth to menopause. Hum. Reprod. 2008, 23, 699–708.
- Faddy, M.J.; Gosden, R.G.; Gougeon, A.; Richardson, S.J.; Nelson, J.F. Accelerated disappearance of ovarian follicles in mid-life: Implications for forecasting menopause. Hum. Reprod. 1992, 7, 1342–1346.
- Cox, E.; Takov, V. Embryology; Ovarian Follicle Development; StatPearls: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532300/ (accessed on 22 May 2022).
- Lebbe, M.; Woodruff, T.K. Involvement of androgens in ovarian health and disease. Mol. Hum. Reprod. 2013, 19, 828–837.
- Franks, S.; Hardy, K. Androgen Action in the Ovary. Front. Endocrinol. 2018, 9, 452.
- Drummond, A.E. The role of steroids in follicular growth. Reprod. Biol. Endocrinol. 2006, 4, 16.
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr. Biol. 2015, 25, R538–R542.
- Coxworth, J.E.; Hawkes, K. Ovarian follicle loss in humans and mice: Lessons from statistical model comparison. Hum. Reprod. 2010, 25, 1796–1805.
- Holton, R.A.; Harris, A.M.; Mukerji, B.; Singh, T.; Dia, F.; Berkowitz, K.M. CHTF18 ensures the quantity and quality of the ovarian reserve. Biol. Reprod. 2020, 103, 24–35.
- Turan, V.; Oktay, K. BRCA-related ATM-mediated DNA double-strand break repair and ovarian aging. Hum. Reprod. Updat. 2020, 26, 43–57.
- Lin, W.; Titus, S.; Moy, F.; Ginsburg, E.S.; Oktay, K. Ovarian Aging in Women with BRCA Germline Mutations. J. Clin. Endocrinol. Metab. 2017, 102, 3839–3847.
- Laven, J.S.E.; Visser, J.A.; Uitterlinden, A.G.; Vermeij, W.P.; Hoeijmakers, J.H.J. Menopause: Genome stability as new paradigm. Maturitas 2016, 92, 15–23.
- Titus, S.; Li, F.; Stobezki, R.; Akula, K.; Unsal, E.; Jeong, K.; Dickler, M.; Robson, M.; Moy, F.; Goswami, S.; et al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013, 5, 172ra21.
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257.
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504.
- Hodges, C.A.; Revenkova, E.; Jessberger, R.; Hassold, T.J.; A Hunt, P. SMC1β-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat. Genet. 2005, 37, 1351–1355.
- Revenkova, E.; Herrmann, K.; Adelfalk, C.; Jessberger, R. Oocyte Cohesin Expression Restricted to Predictyate Stages Provides Full Fertility and Prevents Aneuploidy. Curr. Biol. 2010, 20, 1529–1533.
- Cheng, J.-M.; Liu, Y.-X. Age-Related Loss of Cohesion: Causes and Effects. Int. J. Mol. Sci. 2017, 18, 1578.
- Herbert, M.; Kalleas, D.; Cooney, D.; Lamb, M.; Lister, L. Meiosis and Maternal Aging: Insights from Aneuploid Oocytes and Trisomy Births. Cold Spring Harb. Perspect. Biol. 2015, 7, a017970.
- Chiang, T.; Duncan, F.E.; Schindler, K.; Schultz, R.M.; Lampson, M.A. Evidence that Weakened Centromere Cohesion Is a Leading Cause of Age-Related Aneuploidy in Oocytes. Curr. Biol. 2010, 20, 1522–1528.
- Ciosk, R.; Shirayama, M.; Shevchenko, A.; Tanaka, T.; Toth, A.; Shevchenko, A.; Nasmyth, K. Cohesin’s Binding to Chromosomes Depends on a Separate Complex Consisting of Scc2 and Scc4 Proteins. Mol. Cell 2000, 5, 243–254.
- Xu, X.; Chen, X.; Zhang, X.; Liu, Y.; Wang, Z.; Wang, P.; Du, Y.; Qin, Y.; Chen, Z.-J. Impaired telomere length and telomerase activity in peripheral blood leukocytes and granulosa cells in patients with biochemical primary ovarian insufficiency. Hum. Reprod. 2017, 32, 201–207.
- Miranda-Furtado, C.L.; Luchiari, H.R.; Chielli Pedroso, D.C.; Kogure, G.S.; Caetano, L.C.; Santana, B.A.; Santana, V.P.; Benetti-Pinto, C.L.; Reis, F.M.; Maciel, M.A.; et al. Skewed X-chromosome inactivation and shorter telomeres associate with idiopathic premature ovarian insufficiency. Fertil. Steril. 2018, 110, 476–485.e1.
- Butts, S.; Riethman, H.; Ratcliffe, S.; Shaunik, A.; Coutifaris, C.; Barnhart, K. Correlation of telomere length and telomerase activity with occult ovarian insufficiency. J. Clin. Endocrinol. Metab. 2009, 94, 4835–4843.
- Wang, S.; Zheng, Y.; Li, J.; Yu, Y.; Zhang, W.; Song, M.; Liu, Z.; Min, Z.; Hu, H.; Jing, Y.; et al. Single-Cell Transcriptomic Atlas of Primate Ovarian Aging. Cell 2020, 180, 585–600.e19.
- Liu, L.; Trimarchi, J.R.; Smith, P.J.S.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46.
- Sasaki, H.; Hamatani, T.; Kamijo, S.; Iwai, M.; Kobanawa, M.; Ogawa, S.; Miyado, K.; Tanaka, M. Impact of Oxidative Stress on Age-Associated Decline in Oocyte Developmental Competence. Front. Endocrinol. 2019, 10, 811.
- Zhang, D.; Keilty, D.; Zhang, Z.; Chian, R. Mitochondria in oocyte aging: Current understanding. Facts Views Vis. ObGyn 2017, 9, 29–38.
- Sallevelt, S.C.E.H.; Dreesen, J.C.F.M.; Drüsedau, M.; Spierts, S.; Coonen, E.; van Tienen, F.H.J.; van Golde, R.J.T.; de Coo, I.F.M.; Geraedts, J.P.M.; Die-Smulders, C.E.M.D.; et al. Preimplantation genetic diagnosis in mitochondrial DNA disorders: Challenge and success. J. Med. Genet. 2013, 50, 125–132.
This entry is offline, you can click here to edit this entry!